Preprocessing and clustering 3k PBMCs 
 ¶
¶
This template demonstrates how to use LaminDB to track data provenance and manage artifacts during a standard single-cell RNA-seq analysis workflow. Adapted from the classic Scanpy PBMC3k tutorial, it has additional code showing how to track the notebook’s execution, save the raw dataset as an artifact, and register the final annotated dataset in your LaminDB instance.
We will process a dataset of 3k PBMCs from a Healthy Donor (freely available from 10x Genomics).
Note: To run examples, if you don’t have a LaminDB instance, create one:
!lamin init --storage ./test-pbmc3k
Show code cell output
→ initialized lamindb: testuser1/test-pbmc3k
from __future__ import annotations
import matplotlib.pyplot as plt
import pandas as pd
import scanpy as sc
import lamindb as ln
Show code cell output
→ connected lamindb: testuser1/test-pbmc3k
Track the notebook’s execution in LaminDB
ln.track()
Show code cell output
→ created Transform('X09ozFVjqQot0000', key='pbmc3k.ipynb'), started new Run('m8JHAOWJFjqanxRQ') at 2026-08-03 16:51:39 UTC
→ notebook imports: lamindb-core==2.9.0 matplotlib==3.11.1 pandas==2.3.3 scanpy==1.12.3
• tip: to identify the notebook across renames, pass the uid: ln.track("X09ozFVjqQot")
Download and unpack the data.
mkdir -p data
cd data
test -f pbmc3k_filtered_gene_bc_matrices.tar.gz || curl https://cf.10xgenomics.com/samples/cell/pbmc3k/pbmc3k_filtered_gene_bc_matrices.tar.gz -o pbmc3k_filtered_gene_bc_matrices.tar.gz
tar -xzf pbmc3k_filtered_gene_bc_matrices.tar.gz
Show code cell output
% Total % Received % Xferd Average Speed Time Time Time Current
Dlo
ad Upload Total Spent Left Speed
0 0 0 0 0 0 0 0 --:--:-- --
:--:-- --:--
:-- 0
100 7443k 100 7443k 0 0 41.7M 0 --:--:-- --:--:-- --:--:-- 42.0M
Read in the count matrix into an AnnData object.
adata = sc.read_10x_mtx(
"data/filtered_gene_bc_matrices/hg19/", # the directory with the `.mtx` file
var_names="gene_symbols", # use gene symbols for the variable names (variables-axis index)
cache=True, # write a cache file for faster subsequent reading
)
adata
Show code cell output
AnnData object with n_obs × n_vars = 2700 × 32738
var: 'gene_ids'
layers: None (.X)
Save the AnnData object with the raw/unprocessed 3k PBMCs in LaminDB¶
ln.Artifact.from_anndata(adata, key="raw_pbmc3k.h5ad").save()
Show code cell output
Artifact(uid='EzLaa4hMEmZqfl2O0000', key='raw_pbmc3k.h5ad', description=None, suffix='.h5ad', kind='dataset', otype='AnnData', size=21594848, hash='olsswItNtrSMTtUsy-zMiA', n_files=None, n_observations=2700, extra_data=None, branch_id=1, created_on_id=1, space_id=1, storage_id=1, run_id=1, schema_id=None, created_by_id=1, created_at=2026-08-03 16:51:40 UTC, is_locked=False, version_tag=None, is_latest=True)
Preprocessing¶
Load the Anndata object with the raw/unprocessed 3k PBMCs from LaminDB¶
adata = ln.Artifact.get(key="raw_pbmc3k.h5ad").load()
Show those genes that yield the highest fraction of counts in each single cell, across all cells.
sc.pl.highest_expr_genes(adata, n_top=20)
Show code cell output
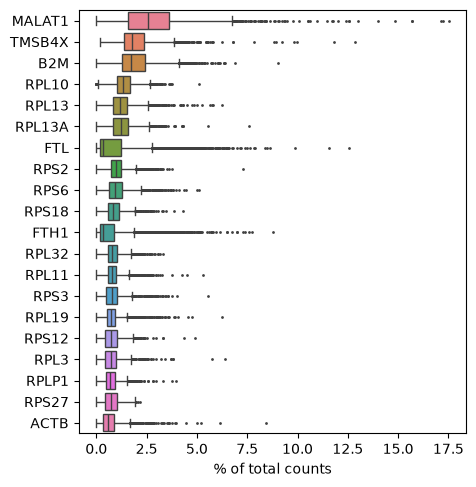
Basic filtering:
sc.pp.filter_cells(adata, min_genes=200)
sc.pp.filter_genes(adata, min_cells=3)
adata
Show code cell output
AnnData object with n_obs × n_vars = 2700 × 13714
obs: 'n_genes'
var: 'gene_ids', 'n_cells'
layers: None (.X)
Let’s assemble some information about mitochondrial genes, which are important for quality control.
# annotate the group of mitochondrial genes as "mt"
adata.var["mt"] = adata.var_names.str.startswith("MT-")
sc.pp.calculate_qc_metrics(adata, qc_vars=["mt"], percent_top=None, log1p=False, inplace=True)
A violin plot of some of the computed quality measures:
the number of genes expressed in the count matrix
the total counts per cell
the percentage of counts in mitochondrial genes
sc.pl.violin(
adata,
["n_genes_by_counts", "total_counts", "pct_counts_mt"],
jitter=0.4,
multi_panel=True,
)
Show code cell output
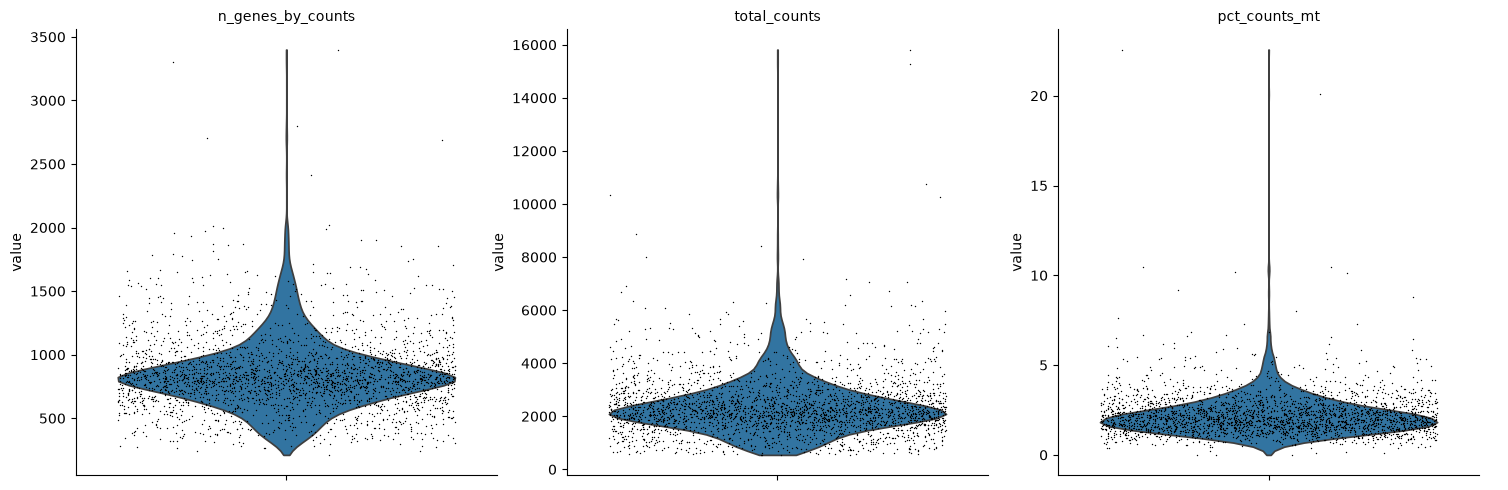
Remove cells that have too many mitochondrial genes expressed or too many total counts. Do the filtering by slicing the AnnData object.
adata = adata[
(adata.obs.n_genes_by_counts < 2500) & (adata.obs.n_genes_by_counts > 200) & (adata.obs.pct_counts_mt < 5),
:,
].copy()
adata.layers["counts"] = adata.X.copy()
adata
Show code cell output
AnnData object with n_obs × n_vars = 2638 × 13714
obs: 'n_genes', 'n_genes_by_counts', 'total_counts', 'total_counts_mt', 'pct_counts_mt'
var: 'gene_ids', 'n_cells', 'mt', 'n_cells_by_counts', 'mean_counts', 'pct_dropout_by_counts', 'total_counts'
layers: None (.X), 'counts'
Total-count normalize (library-size correct) the data matrix $\mathbf{X}$ to 10,000 reads per cell, so that counts become comparable among cells.
sc.pp.normalize_total(adata, target_sum=1e4)
Logarithmize the data.
sc.pp.log1p(adata)
Identify highly-variable genes.
sc.pp.highly_variable_genes(
adata,
layer="counts",
n_top_genes=2000,
min_mean=0.0125,
max_mean=3,
min_disp=0.5,
flavor="seurat_v3",
)
sc.pl.highly_variable_genes(adata)
Show code cell output
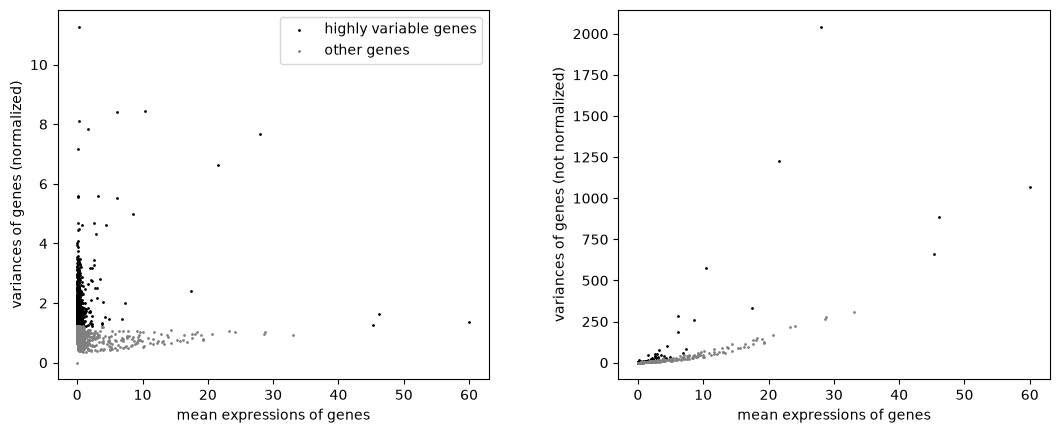
Scale each gene to unit variance. Clip values exceeding standard deviation 10.
adata.layers["scaled"] = adata.X.toarray(order='C')
sc.pp.regress_out(adata, ["total_counts", "pct_counts_mt"], layer="scaled")
sc.pp.scale(adata, max_value=10, layer="scaled")
Show code cell output
/opt/hostedtoolcache/Python/3.12.13/x64/lib/python3.12/site-packages/scanpy/preprocessing/_simple.py:660: NumbaPerformanceWarning: '@' is faster on contiguous arrays, called on (Array(float64, 1, 'A', False, aligned=True), Array(float64, 2, 'C', False, aligned=True))
data[i] -= regressor[i] @ coeff
/opt/hostedtoolcache/Python/3.12.13/x64/lib/python3.12/site-packages/scanpy/preprocessing/_simple.py:660: NumbaPerformanceWarning: '@' is faster on contiguous arrays, called on (Array(float64, 1, 'A', False, aligned=True), Array(float64, 2, 'C', False, aligned=True))
data[i] -= regressor[i] @ coeff
Principal component analysis¶
Reduce the dimensionality of the data by running principal component analysis (PCA), which reveals the main axes of variation and denoises the data.
sc.pp.pca(adata, layer="scaled", svd_solver="arpack")
Let us inspect the contribution of single PCs to the total variance in the data. This gives us information about how many PCs we should consider in order to compute the neighborhood relations of cells, e.g. used in the clustering function sc.tl.louvain() or tSNE sc.tl.tsne().
sc.pl.pca_variance_ratio(adata, n_pcs=20)
Show code cell output
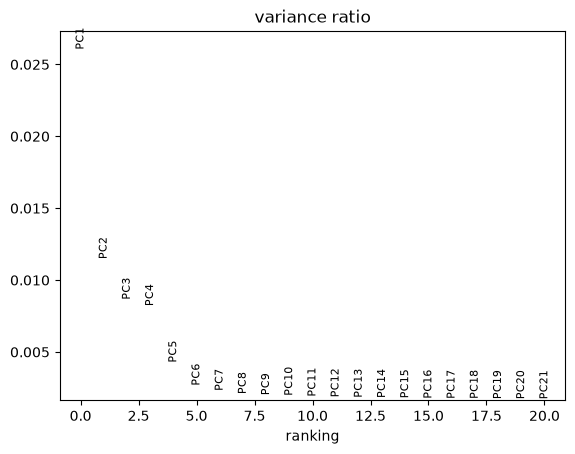
Computing the neighborhood graph¶
Compute the neighborhood graph of cells using the PCA representation of the data matrix.
sc.pp.neighbors(adata, n_neighbors=10, n_pcs=40)
Clustering the neighborhood graph¶
sc.tl.leiden(
adata,
resolution=0.7,
random_state=0,
flavor="igraph",
n_iterations=2,
directed=False,
)
adata.obs["leiden"] = adata.obs["leiden"].copy()
adata.uns["leiden"] = adata.uns["leiden"].copy()
Embedding the neighborhood graph in a UMAP¶
sc.tl.paga(adata)
sc.pl.paga(adata, plot=False) # remove `plot=False` if you want to see the coarse-grained graph
sc.tl.umap(adata, init_pos='paga')
sc.tl.umap(adata)
adata.obsm["X_umap"] = adata.obsm["X_umap"].copy()
sc.pl.umap(adata, color=["leiden", "CD14", "NKG7"])
Show code cell output
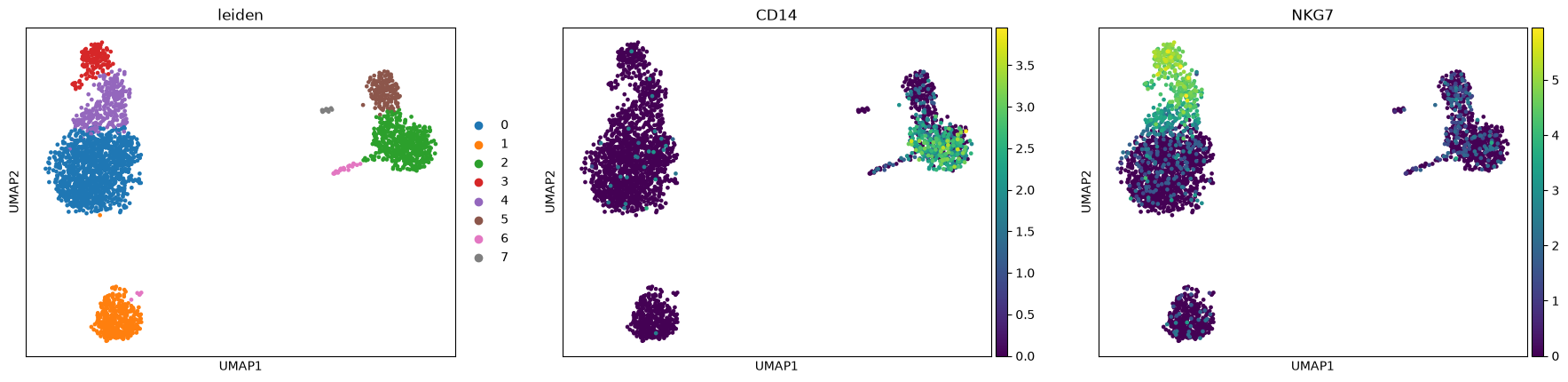
Finding marker genes¶
Let us compute a ranking for the highly differential genes in each cluster.
sc.tl.rank_genes_groups(adata, "leiden", mask_var="highly_variable", method="wilcoxon")
sc.pl.rank_genes_groups(adata, n_genes=25, sharey=False)
Show code cell output
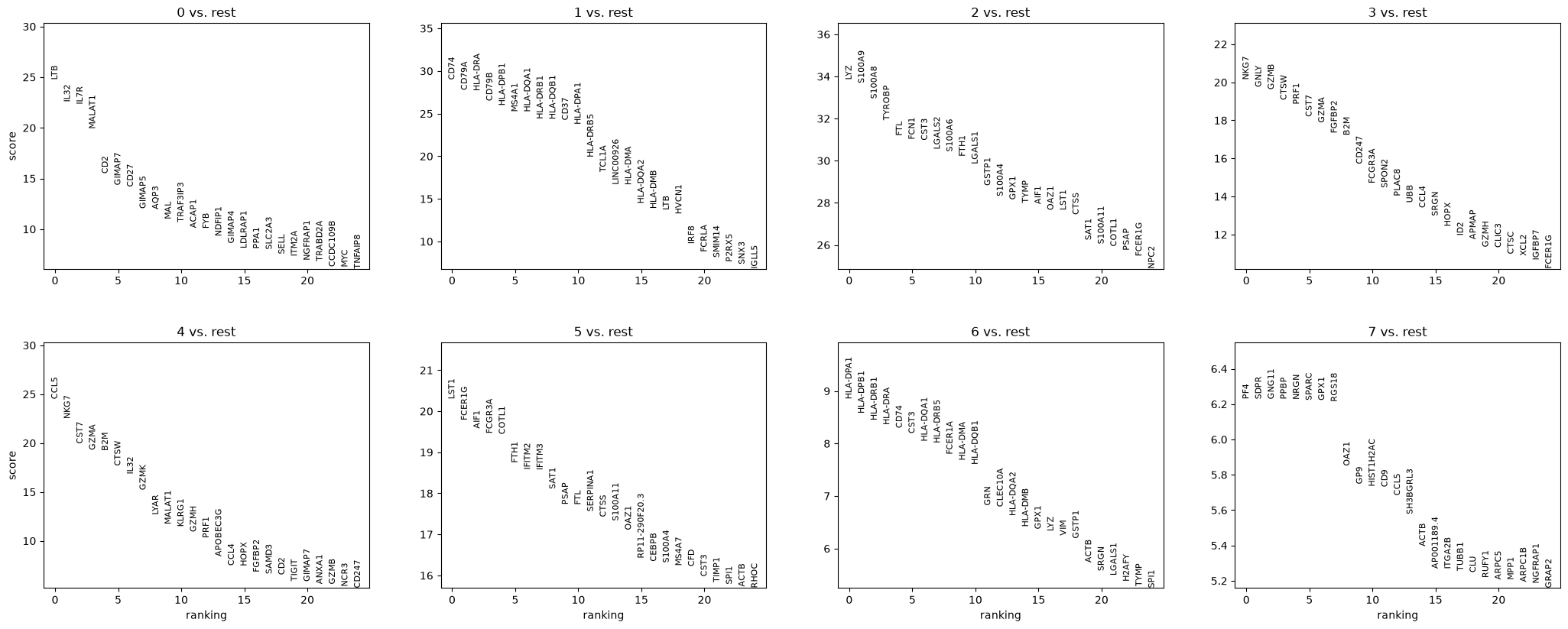
Most of these genes are found in the highly expressed genes with notable exceptions like CD8A (see the below dotplot).
Leiden Group |
Markers |
Cell Type |
|---|---|---|
0 |
IL7R |
CD4 T cells |
1 |
CD14, LYZ |
CD14+ Monocytes |
2 |
MS4A1 |
B cells |
3 |
CD8A |
CD8 T cells |
4 |
GNLY, NKG7 |
NK cells |
5 |
FCGR3A, MS4A7 |
FCGR3A+ Monocytes |
6 |
FCER1A, CST3 |
Dendritic Cells |
7 |
PPBP |
Megakaryocytes |
Let us also define a list of marker genes for later reference.
marker_genes = [
*["IL7R", "CD79A", "MS4A1", "CD8A", "CD8B", "LYZ", "CD14"],
*["LGALS3", "S100A8", "GNLY", "NKG7", "KLRB1"],
*["FCGR3A", "MS4A7", "FCER1A", "CST3", "PPBP"],
]
Actually mark the cell types.
new_cluster_names = [
"CD4 T",
"B",
"CD14+ Monocytes",
"NK",
"CD8 T",
"FCGR3A+ Monocytes",
"Dendritic",
"Megakaryocytes",
]
adata.rename_categories("leiden", new_cluster_names)
adata.obs['cell_type']=adata.obs['leiden']
adata
Show code cell output
AnnData object with n_obs × n_vars = 2638 × 13714
obs: 'n_genes', 'n_genes_by_counts', 'total_counts', 'total_counts_mt', 'pct_counts_mt', 'leiden', 'cell_type'
var: 'gene_ids', 'n_cells', 'mt', 'n_cells_by_counts', 'mean_counts', 'pct_dropout_by_counts', 'total_counts', 'highly_variable', 'highly_variable_rank', 'means', 'variances', 'variances_norm', 'mean', 'std'
uns: 'log1p', 'hvg', 'pca', 'neighbors', 'leiden', 'paga', 'leiden_sizes', 'umap', 'leiden_colors', 'rank_genes_groups'
obsm: 'X_pca', 'X_umap'
varm: 'PCs'
obsp: 'distances', 'connectivities'
layers: None (.X), 'counts', 'scaled'
Visualize the cell types in a UMAP
sc.pl.umap(adata, color="cell_type", legend_loc="on data", title="", frameon=False)
Show code cell output
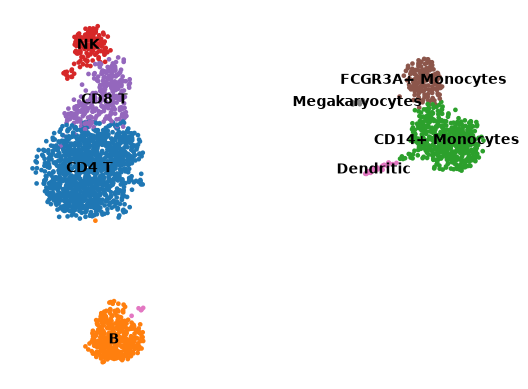
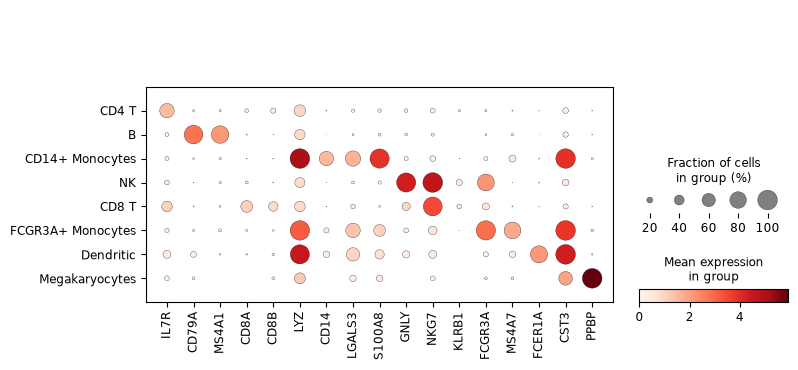
Now that we annotated the cell types, let us visualize the marker genes.
sc.pl.dotplot(adata, marker_genes, groupby="cell_type")
Show code cell output
Save the resulting AnnData object in LaminDB¶
ln.Artifact.from_anndata(adata, key="result_pbmc3k.h5ad").save()
Show code cell output
Artifact(uid='OIxoWqhkpVMU35WM0000', key='result_pbmc3k.h5ad', description=None, suffix='.h5ad', kind='dataset', otype='AnnData', size=190921732, hash='zF7d2Z6wGilsrzby-WCF_R', n_files=None, n_observations=2638, extra_data=None, branch_id=1, created_on_id=1, space_id=1, storage_id=1, run_id=1, schema_id=None, created_by_id=1, created_at=2026-08-03 16:52:28 UTC, is_locked=False, version_tag=None, is_latest=True)
ln.finish() marks the successful completion of the current script or notebook run in LaminDB, finalizing its execution state and saving the source code to the database for reproducibility.
ln.finish()
Show code cell output
→ finished Run('m8JHAOWJFjqanxRQ') after 51s at 2026-08-03 16:52:30 UTC