CELLxGENE 
 ¶
¶
CZ CELLxGENE hosts one of the largest standardized collections of scRNA-seq data - LaminDB provides a streamlined interface to query and load it.
You can use the CELLxGENE data in two ways:
Query collections of
AnnDataobjects.Query slices from a single concatenated dataset without downloading everything via TileDB-SOMA.
To build similar data assets in-house:
See the transfer guide to zero-copy data to your own LaminDB instance.
See the scRNA guide to create a growing, standardized & versioned scRNA-seq dataset collection.
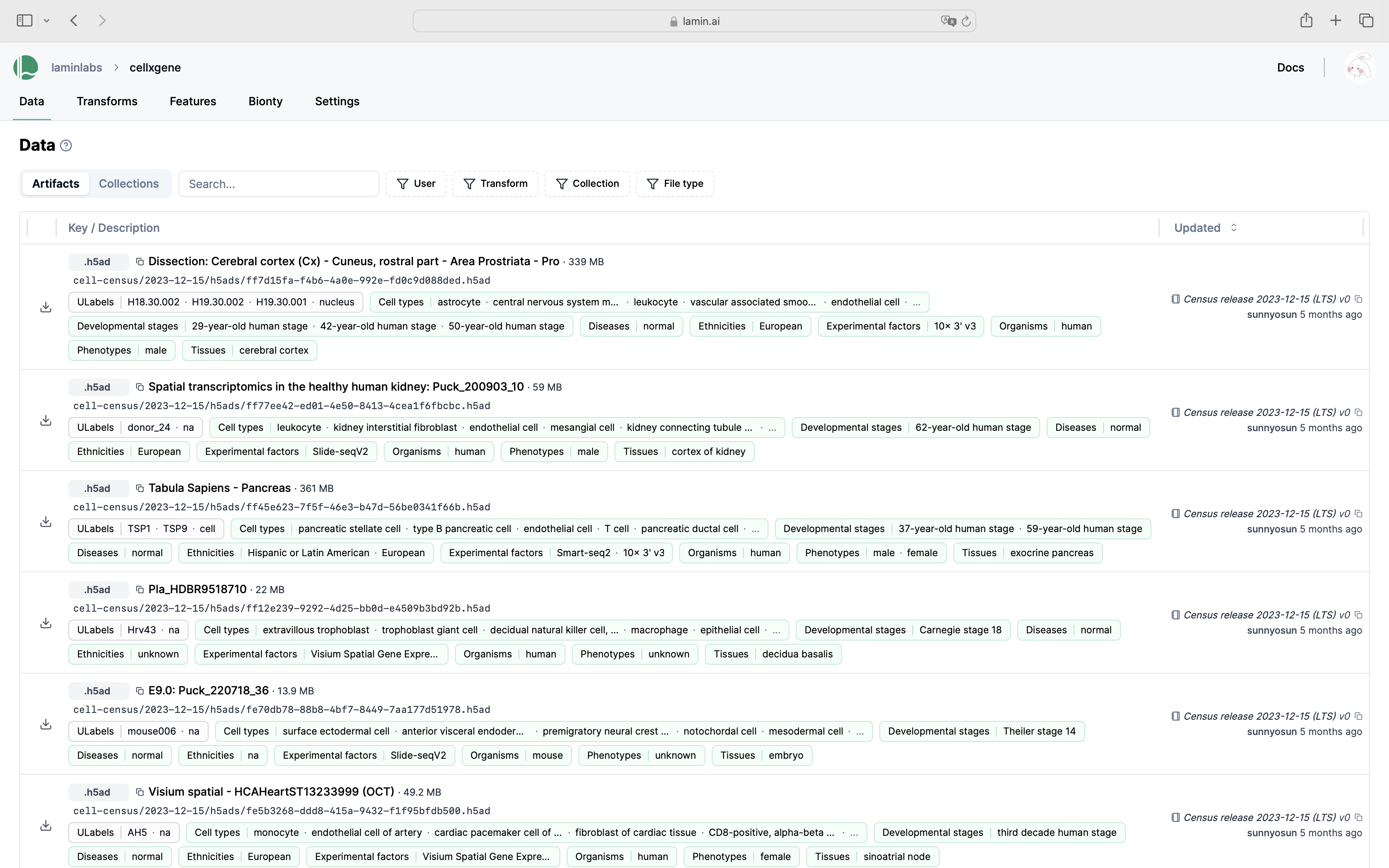
Show me a screenshot
import lamindb as ln
Create the central query object for the public laminlabs/cellxgene instance:
db = ln.DB("laminlabs/cellxgene")
Show code cell output
! using anonymous user (to identify, call: lamin login)
Query for individual datasets¶
Every individual dataset in CELLxGENE is an .h5ad file that is stored as an artifact in LaminDB. Here is an exemplary query:
users = db.User.lookup()
cell_types = db.bionty.CellType.lookup()
db.Artifact.filter(
suffix=".h5ad",
description__contains="immune",
size__gt=1e9, # size > 1GB
cell_types__name__in=["B cell", "T cell"], # cell types measured in AnnData
created_by=users.sunnyosun, # created by a specific user
).order_by("created_at").to_dataframe(
include=["cell_types__name", "created_by__handle"] # join with additional info
).head()
Show code cell output
! truncated query result to limit=20 Artifact objects
| uid | key | cell_types__name | created_by__handle | |
|---|---|---|---|---|
| id | ||||
| 879 | BCutg5cxmqLmy2Z5SS8J | cell-census/2023-07-25/h5ads/01ad3cd7-3929-465... | {natural killer cell, B cell, classical monocy... | sunnyosun |
| 1106 | 3xdOASXuAxxJtSchJO3D | cell-census/2023-07-25/h5ads/48101fa2-1a63-451... | {innate lymphoid cell, plasma cell, group 2 in... | sunnyosun |
| 1174 | wt7eD72sTzwL3rfYaZr2 | cell-census/2023-07-25/h5ads/58b01044-c5e5-4b0... | {natural killer cell, stromal cell, B cell, in... | sunnyosun |
| 1377 | znTBqWgfYgFlLjdQ6Ba7 | cell-census/2023-07-25/h5ads/9dbab10c-118d-496... | {natural killer cell, CD14-positive, CD16-posi... | sunnyosun |
| 1438 | N849OuhsgqIerohvFiOv | cell-census/2023-07-25/h5ads/ac2fea99-ce08-4fc... | {natural killer cell, megakaryocyte, B cell, i... | sunnyosun |
What happens under the hood?
As you saw from inspecting Artifact, Artifact.cell_types relates artifacts with bionty.CellType.
The expression cell_types__name__in performs the join of the underlying registries and matches bionty.CellType.name to ["B cell", "T cell"].
Similar for created_by, which relates artifacts with User.
Slice an individual dataset¶
Let’s look at a CELLxGENE artifact and show its metadata using .describe().
artifact = db.Artifact.get(description="Mature kidney dataset: immune")
artifact.describe()
Show code cell output
Artifact: cell-census/2025-11-08/h5ads/20d87640-4be8-487f-93d4-dce38378d00f.h5ad (2025-11-08) | description: Mature kidney dataset: immune ├── uid: WwmBIhBNLTlRcSoBDt78 run: 7FgSsR6 (annotate-register-new-release.py) │ kind: None otype: AnnData │ hash: I66HxDXLtdy_NzTmgv2Mrg size: 42.0 MB │ branch: main space: all │ created_at: 2026-02-17 13:37:22 UTC created_by: zethson │ n_observations: 7803.0 schema: CELLxGENE AnnData of ontology_id ├── storage/path: s3://cellxgene-data-public/cell-census/2025-11-08/h5ads/20d87640-4be8-487f-93d4-dce38378d00f.h5ad ├── Dataset features │ ├── obs (11.0) │ │ assay_ontology_term_id bionty.ExperimentalFactor.ontology… EFO:0009899 │ │ cell_type_ontology_term_id bionty.CellType.ontology_id CL:0000097, CL:0000236, CL:0000451, CL… │ │ development_stage_ontology_t… bionty.DevelopmentalStage.ontology… HsapDv:0000096, HsapDv:0000098, HsapDv… │ │ disease_ontology_term_id bionty.Disease.ontology_id PATO:0000461 │ │ donor_id str │ │ is_primary_data ULabel │ │ self_reported_ethnicity_onto… bionty.Ethnicity.ontology_id unknown │ │ sex_ontology_term_id bionty.Phenotype.ontology_id PATO:0000383, PATO:0000384 │ │ suspension_type ULabel cell │ │ tissue_ontology_term_id bionty.Tissue.ontology_id|bionty.C… UBERON:0000362, UBERON:0001224, UBERON… │ │ tissue_type ULabel tissue │ ├── uns (1.0) │ │ organism_ontology_term_id bionty.Organism.ontology_id NCBITaxon:9606 │ └── var (2.0) │ feature_is_filtered bool │ var_index bionty.Gene.ensembl_gene_id[source… └── Labels └── .recreating_runs Run 2026-02-17 13:43:36.195894+00:00, 2026… .ulabels ULabel cell, tissue .organisms bionty.Organism human .tissues bionty.Tissue kidney blood vessel, renal pelvis, cor… .cell_types bionty.CellType mast cell, B cell, dendritic cell, nat… .diseases bionty.Disease normal .phenotypes bionty.Phenotype female, male .experimental_factors bionty.ExperimentalFactor 10x 3' v2 .developmental_stages bionty.DevelopmentalStage 2-year-old stage, 4-year-old stage, 12… .ethnicities bionty.Ethnicity unknown
More ways of accessing metadata
Access just features:
artifact.features
Or get values associated with features:
artifact.features.get_values()
To query & load a slice of the array data, you have several options:
Cache the artifact on disk and return the path to the cached data like:
artifact.cache() -> PathCache & load the entire artifact into memory via
artifact.load() -> AnnDataStream the array using a (cloud-backed) accessor
artifact.open() -> AnnDataAccessor
All of these option run much faster in the AWS us-west-2 data center.
Cache:
cache_path = artifact.cache()
cache_path
Show code cell output
PosixUPath('/home/runner/.cache/lamindb/cellxgene-data-public/cell-census/2025-11-08/h5ads/20d87640-4be8-487f-93d4-dce38378d00f.h5ad')
Cache & load:
adata = artifact.load()
adata
Show code cell output
AnnData object with n_obs × n_vars = 7803 × 32088
obs: 'donor_id', 'donor_age', 'self_reported_ethnicity_ontology_term_id', 'sample_uuid', 'tissue_ontology_term_id', 'development_stage_ontology_term_id', 'suspension_uuid', 'suspension_type', 'library_uuid', 'assay_ontology_term_id', 'mapped_reference_annotation', 'is_primary_data', 'cell_type_ontology_term_id', 'author_cell_type', 'disease_ontology_term_id', 'reported_diseases', 'sex_ontology_term_id', 'compartment', 'Experiment', 'Project', 'tissue_type', 'cell_type', 'assay', 'disease', 'sex', 'tissue', 'self_reported_ethnicity', 'development_stage', 'observation_joinid'
var: 'feature_is_filtered', 'feature_name', 'feature_reference', 'feature_biotype', 'feature_length', 'feature_type'
uns: 'citation', 'default_embedding', 'organism', 'organism_ontology_term_id', 'schema_reference', 'schema_version', 'title'
obsm: 'X_umap'
layers: None (.X)
Now we have an AnnData object, which stores observation annotations matching our artifact-level query in the .obs slot, and we can re-use almost the same query on the array-level.
tissues = db.bionty.Tissue.lookup()
suspension_types = db.ULabel.filter(type__name="SuspensionType").lookup()
experimental_factors = db.bionty.ExperimentalFactor.lookup()
adata_slice = adata[
adata.obs.cell_type.isin(
[cell_types.dendritic_cell.name, cell_types.neutrophil.name]
)
& (adata.obs.tissue == tissues.kidney.name)
& (adata.obs.suspension_type == suspension_types.cell.name)
& (adata.obs.assay == experimental_factors.ln_10x_3_v2.name)
]
adata_slice
Show code cell output
View of AnnData object with n_obs × n_vars = 199 × 32088
obs: 'donor_id', 'donor_age', 'self_reported_ethnicity_ontology_term_id', 'sample_uuid', 'tissue_ontology_term_id', 'development_stage_ontology_term_id', 'suspension_uuid', 'suspension_type', 'library_uuid', 'assay_ontology_term_id', 'mapped_reference_annotation', 'is_primary_data', 'cell_type_ontology_term_id', 'author_cell_type', 'disease_ontology_term_id', 'reported_diseases', 'sex_ontology_term_id', 'compartment', 'Experiment', 'Project', 'tissue_type', 'cell_type', 'assay', 'disease', 'sex', 'tissue', 'self_reported_ethnicity', 'development_stage', 'observation_joinid'
var: 'feature_is_filtered', 'feature_name', 'feature_reference', 'feature_biotype', 'feature_length', 'feature_type'
uns: 'citation', 'default_embedding', 'organism', 'organism_ontology_term_id', 'schema_reference', 'schema_version', 'title'
obsm: 'X_umap'
layers: None (.X)
Stream, slice and load the slice into memory:
adata_backed = artifact.open()
We now have an AnnDataAccessor object, which behaves much like an AnnData, and slicing looks similar to the query above.
See the slicing operation:
adata_backed_slice = adata_backed[
adata_backed.obs.cell_type.isin(
[cell_types.dendritic_cell.name, cell_types.neutrophil.name]
)
& (adata_backed.obs.tissue == tissues.kidney.name)
& (adata_backed.obs.suspension_type == suspension_types.cell.name)
& (adata_backed.obs.assay == experimental_factors.ln_10x_3_v2.name)
]
adata_backed_slice.to_memory()
Show code cell output
AnnData object with n_obs × n_vars = 199 × 32088
obs: 'donor_id', 'donor_age', 'self_reported_ethnicity_ontology_term_id', 'sample_uuid', 'tissue_ontology_term_id', 'development_stage_ontology_term_id', 'suspension_uuid', 'suspension_type', 'library_uuid', 'assay_ontology_term_id', 'mapped_reference_annotation', 'is_primary_data', 'cell_type_ontology_term_id', 'author_cell_type', 'disease_ontology_term_id', 'reported_diseases', 'sex_ontology_term_id', 'compartment', 'Experiment', 'Project', 'tissue_type', 'cell_type', 'assay', 'disease', 'sex', 'tissue', 'self_reported_ethnicity', 'development_stage', 'observation_joinid'
var: 'feature_is_filtered', 'feature_name', 'feature_reference', 'feature_biotype', 'feature_length', 'feature_type'
uns: 'citation', 'default_embedding', 'organism', 'organism_ontology_term_id', 'schema_reference', 'schema_version', 'title'
obsm: 'X_umap'
layers: None (.X)
adata_backed.close()
Query collections of datasets¶
Let’s search collections from CELLxGENE within the 2025-01-30 release: https://lamin.ai/laminlabs/cellxgene/collections, and then pick a top hit:
It’s a Science paper and we can find more information on it using the DOI or CELLxGENE collection id. There are multiple versions of this collection.
collection = db.Collection.get("quQDnLsMLkP3JRsC8gp5")
collection
Show code cell output
Collection(uid='quQDnLsMLkP3JRsC8gp5', key='Single-cell transcriptomic atlas for adult human retina', description='10.1016/j.xgen.2023.100298', hash='_bg3b6SweW6v1TJX7NgHCw', reference='af893e86-8e9f-41f1-a474-ef05359b1fb7', reference_type='CELLxGENE Collection ID', meta_artifact=None, branch_id=1, created_on_id=1, space_id=1, created_by_id=8, run_id='44', schema_id=None, created_at=2025-08-04 14:53:38 UTC, is_locked=False, version_tag='2025-01-30 00:00:00 ', is_latest=False)
collection.versions.to_dataframe()
Show code cell output
| uid | key | description | hash | reference | reference_type | version_tag | is_latest | is_locked | created_at | branch_id | created_on_id | space_id | created_by_id | run_id | schema_id | meta_artifact_id | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| id | |||||||||||||||||
| 901 | quQDnLsMLkP3JRsC8gp6 | Single-cell transcriptomic atlas for adult hum... | 10.1016/j.xgen.2023.100298 | DvrNP9mC808eREtN70Rh6A | af893e86-8e9f-41f1-a474-ef05359b1fb7 | CELLxGENE Collection ID | 2025-11-08 | True | False | 2026-02-17 13:40:05.630988+00:00 | 1 | 1 | 1 | 8 | 71 | None | None |
| 767 | quQDnLsMLkP3JRsC8gp5 | Single-cell transcriptomic atlas for adult hum... | 10.1016/j.xgen.2023.100298 | _bg3b6SweW6v1TJX7NgHCw | af893e86-8e9f-41f1-a474-ef05359b1fb7 | CELLxGENE Collection ID | 2025-01-30 | False | False | 2025-08-04 14:53:38.098405+00:00 | 1 | 1 | 1 | 8 | 44 | None | None |
| 606 | quQDnLsMLkP3JRsC8gp4 | Single-cell transcriptomic atlas for adult hum... | 10.1016/j.xgen.2023.100298 | NIo8G6_reJTEqMzW2nMc | af893e86-8e9f-41f1-a474-ef05359b1fb7 | CELLxGENE Collection ID | 2024-07-01 | False | False | 2024-07-16 12:21:51.449109+00:00 | 1 | 1 | 1 | 1 | 27 | None | None |
| 291 | quQDnLsMLkP3JRsCJNGB | Single-cell transcriptomic atlas for adult hum... | 10.1016/j.xgen.2023.100298 | FsD52kpR7dF2h78-P3ka | af893e86-8e9f-41f1-a474-ef05359b1fb7 | CELLxGENE Collection ID | 2023-12-15 | False | False | 2024-01-11 13:41:01.880382+00:00 | 1 | 1 | 1 | 1 | 22 | None | None |
| 134 | quQDnLsMLkP3JRsC6WWz | Single-cell transcriptomic atlas for adult hum... | 10.1016/j.xgen.2023.100298 | xhfSShX8lypXPx00zevx | af893e86-8e9f-41f1-a474-ef05359b1fb7 | CELLxGENE Collection ID | 2023-07-25 | False | False | 2024-01-08 12:22:12.891930+00:00 | 1 | 1 | 1 | 1 | None | None | None |
The collection groups artifacts.
collection.artifacts.to_dataframe()
Show code cell output
| uid | key | description | suffix | kind | otype | size | hash | n_files | n_observations | ... | is_latest | is_locked | created_at | branch_id | created_on_id | space_id | storage_id | run_id | schema_id | created_by_id | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| id | |||||||||||||||||||||
| 5156 | 80xlsVmayPPBCCEZ7aBd | cell-census/2025-01-30/h5ads/ed419b4e-db9b-40f... | Non-neuronal cells in human retina | .h5ad | None | AnnData | 1395529802 | RRN8NmTfDVqYChkbH9P1Mw | None | 18011.0 | ... | False | False | 2025-07-30 09:51:11.825708+00:00 | 1 | 1 | 1 | 2 | 43 | 1569 | 8 |
| 4755 | Ce4Mqe4X2vUhwkwnh5YR | cell-census/2025-01-30/h5ads/aad97cb5-f375-45e... | Retinal ganglion cells in human retina | .h5ad | None | AnnData | 784575110 | 3F_RaFf5KXsu_IJ64ZuLzw | None | 11617.0 | ... | False | False | 2025-07-30 09:51:11.140803+00:00 | 1 | 1 | 1 | 2 | 43 | 1569 | 8 |
| 4562 | 1OyQQLNfu1nzvVADODNE | cell-census/2025-01-30/h5ads/8f10185b-e0b3-46a... | Bipolar cells in human retina | .h5ad | None | AnnData | 3075769786 | tJgT6hNoEP8pF8E26Jtc0g | None | 53040.0 | ... | False | False | 2025-07-30 09:51:10.819182+00:00 | 1 | 1 | 1 | 2 | 43 | 1569 | 8 |
| 4044 | QpuY5RsGTBBMN61QGY4u | cell-census/2025-01-30/h5ads/359f7af4-87d4-411... | Amacrine cells in human retina | .h5ad | None | AnnData | 3382157940 | -wVd9eb1ZIvMQxbKU3AFng | None | 56507.0 | ... | False | False | 2025-07-30 09:51:09.958055+00:00 | 1 | 1 | 1 | 2 | 43 | 1569 | 8 |
| 3819 | GA2BXWwoJlcRfzNp3iyR | cell-census/2025-01-30/h5ads/11ef37ee-2173-458... | Horizontal cells in human retina | .h5ad | None | AnnData | 404983068 | WReD9Id8iqAiGnKfysjk6Q | None | 7348.0 | ... | False | False | 2025-07-30 09:51:09.581354+00:00 | 1 | 1 | 1 | 2 | 43 | 1569 | 8 |
| 3703 | wYiUe9hn4TJijpoX90Ms | cell-census/2025-01-30/h5ads/0129dbd9-a7d3-4f6... | All major cell types in adult human retina | .h5ad | None | AnnData | 18955176484 | jCYVCnU2ZEkiBl396t_cJA | None | 244474.0 | ... | False | False | 2025-07-30 09:51:09.357674+00:00 | 1 | 1 | 1 | 2 | 43 | 1569 | 8 |
| 3700 | Oc6ANFJ0FgOW1B70mNIr | cell-census/2025-01-30/h5ads/00e5dedd-b9b7-43b... | Photoreceptor cells in human retina (rod cells... | .h5ad | None | AnnData | 1347305774 | R9kY-9aSvG9pXhwEX-ZjLw | None | 21422.0 | ... | False | False | 2025-07-30 09:51:09.348148+00:00 | 1 | 1 | 1 | 2 | 43 | 1569 | 8 |
7 rows × 22 columns
Let’s now look at the collection that corresponds to a cellxgene-census release of .h5ad artifacts.
collection = db.Collection.get(key="cellxgene-census", version="2025-11-08")
collection
Show code cell output
Collection(uid='dMyEX3NTfKOEYXyMKDDA', key='cellxgene-census', description=None, hash='5pr6rINICyOqUGmbihW4Sg', reference=None, reference_type=None, meta_artifact=None, branch_id=1, created_on_id=1, space_id=1, created_by_id=8, run_id='72', schema_id=None, created_at=2026-02-17 13:47:54 UTC, is_locked=False, version_tag='2025-11-08 00:00:00 ', is_latest=True)
You can query across artifacts by arbitrary metadata combinations, for instance:
organisms = db.bionty.Organism.lookup()
experimental_factors = db.bionty.ExperimentalFactor.lookup()
tissues = db.bionty.Tissue.lookup()
suspension_types = db.ULabel.filter(type__name="SuspensionType").lookup()
features = db.Feature.lookup(
return_field="name"
) # here we choose to return .name directly
assays = db.bionty.ExperimentalFactor.lookup(return_field="name")
query = collection.artifacts.filter(
organisms=organisms.human,
cell_types__in=[cell_types.dendritic_cell, cell_types.neutrophil],
tissues=tissues.kidney,
ulabels=suspension_types.cell,
experimental_factors=experimental_factors.ln_10x_3_v2,
)
query = query.order_by("size")
query.to_dataframe().head()
Show code cell output
| uid | key | description | suffix | kind | otype | size | hash | n_files | n_observations | ... | is_latest | is_locked | created_at | branch_id | created_on_id | space_id | storage_id | run_id | schema_id | created_by_id | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| id | |||||||||||||||||||||
| 5583 | WwmBIhBNLTlRcSoBDt78 | cell-census/2025-11-08/h5ads/20d87640-4be8-487... | Mature kidney dataset: immune | .h5ad | None | AnnData | 44088792 | I66HxDXLtdy_NzTmgv2Mrg | None | 7803.0 | ... | True | False | 2026-02-17 13:37:22.694797+00:00 | 1 | 1 | 1 | 2 | 71 | 1573 | 8 |
| 5674 | gHlQ5Muwu3G9pvFCx3xA | cell-census/2025-11-08/h5ads/2d31c0ca-0233-41c... | Fetal kidney dataset: immune | .h5ad | None | AnnData | 62986868 | JXMh5h36W8a59P3gra1z-Q | None | 6847.0 | ... | True | False | 2026-02-17 13:37:31.074854+00:00 | 1 | 1 | 1 | 2 | 71 | 1573 | 8 |
| 6480 | P4Oai3OLGAzRwoicHfLO | cell-census/2025-11-08/h5ads/9ea768a2-87ab-46b... | Mature kidney dataset: full | .h5ad | None | AnnData | 191551038 | 960YYJ3XZ2ugiI-8ddY0_w | None | 40268.0 | ... | True | False | 2026-02-17 13:38:48.839287+00:00 | 1 | 1 | 1 | 2 | 71 | 1573 | 8 |
| 5459 | DSpevwaIl5E2jIWHbui6 | cell-census/2025-11-08/h5ads/105c7dad-0468-462... | mature | .h5ad | None | AnnData | 231809419 | 2YuQK3mO2AF_i7r2fvxDzQ | None | 40268.0 | ... | True | False | 2026-02-17 13:37:11.461274+00:00 | 1 | 1 | 1 | 2 | 71 | 1573 | 8 |
| 6868 | 11HQaMeIUaOwyHoOkqqO | cell-census/2025-11-08/h5ads/d7dcfd8f-2ee7-438... | Fetal kidney dataset: full | .h5ad | None | AnnData | 338566028 | 7f8EqcMXKcoWA-lq9ijX_g | None | 27197.0 | ... | True | False | 2026-02-17 13:39:26.587260+00:00 | 1 | 1 | 1 | 2 | 71 | 1573 | 8 |
5 rows × 22 columns
Slice a concatenated array¶
Let us now use the concatenated version of the Census collection: a tiledbsoma array that concatenates all AnnData arrays present in the collection we just explored. Slicing tiledbsoma works similar to slicing DataFrame or AnnData.
value_filter = (
f'{features.tissue} == "{tissues.brain.name}" and {features.cell_type} in'
f' ["{cell_types.microglial_cell.name}", "{cell_types.neuron.name}"] and'
f' {features.suspension_type} == "{suspension_types.cell.name}" and {features.assay} =='
f' "{assays.ln_10x_3_v3}"'
)
value_filter
Show code cell output
'tissue == "brain" and cell_type in ["microglial cell", "neuron"] and suspension_type == "cell" and assay == "10x 3\' v3"'
Query for the tiledbsoma array store that contains all concatenated expression data. It’s a new dataset produced by concatenating all AnnData-like artifacts in the Census collection.
census_artifact = db.Artifact.get(key="cell-census/2025-11-08/soma")
Run the slicing operation.
human = "homo_sapiens" # subset to human data
# open the array store for queries
with census_artifact.open() as store:
# read SOMADataFrame as a slice
cell_metadata = store["census_data"][human].obs.read(value_filter=value_filter)
# concatenate results to pyarrow.Table
cell_metadata = cell_metadata.concat()
# convert to pandas.DataFrame
cell_metadata = cell_metadata.to_pandas()
cell_metadata.head()
Show code cell output
| soma_joinid | dataset_id | assay | assay_ontology_term_id | cell_type | cell_type_ontology_term_id | development_stage | development_stage_ontology_term_id | disease | disease_ontology_term_id | ... | tissue | tissue_ontology_term_id | tissue_type | tissue_general | tissue_general_ontology_term_id | raw_sum | nnz | raw_mean_nnz | raw_variance_nnz | n_measured_vars | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | 58774623 | 1b52ee22-c71a-4a0f-a4d2-c1be36d0b82a | 10x 3' v3 | EFO:0009922 | neuron | CL:0000540 | Carnegie stage 18 | HsapDv:0000025 | normal | PATO:0000461 | ... | brain | UBERON:0000955 | tissue | brain | UBERON:0000955 | 8225.0 | 3612 | 2.277132 | 27.249956 | 58226 |
| 1 | 58774624 | 1b52ee22-c71a-4a0f-a4d2-c1be36d0b82a | 10x 3' v3 | EFO:0009922 | neuron | CL:0000540 | Carnegie stage 18 | HsapDv:0000025 | normal | PATO:0000461 | ... | brain | UBERON:0000955 | tissue | brain | UBERON:0000955 | 6507.0 | 2901 | 2.243020 | 41.892300 | 58226 |
| 2 | 58774625 | 1b52ee22-c71a-4a0f-a4d2-c1be36d0b82a | 10x 3' v3 | EFO:0009922 | neuron | CL:0000540 | Carnegie stage 18 | HsapDv:0000025 | normal | PATO:0000461 | ... | brain | UBERON:0000955 | tissue | brain | UBERON:0000955 | 10495.0 | 3850 | 2.725974 | 75.410211 | 58226 |
| 3 | 58774626 | 1b52ee22-c71a-4a0f-a4d2-c1be36d0b82a | 10x 3' v3 | EFO:0009922 | neuron | CL:0000540 | Carnegie stage 18 | HsapDv:0000025 | normal | PATO:0000461 | ... | brain | UBERON:0000955 | tissue | brain | UBERON:0000955 | 9971.0 | 3855 | 2.586511 | 44.339621 | 58226 |
| 4 | 58774627 | 1b52ee22-c71a-4a0f-a4d2-c1be36d0b82a | 10x 3' v3 | EFO:0009922 | neuron | CL:0000540 | Carnegie stage 18 | HsapDv:0000025 | normal | PATO:0000461 | ... | brain | UBERON:0000955 | tissue | brain | UBERON:0000955 | 13675.0 | 4726 | 2.893567 | 53.232056 | 58226 |
5 rows × 28 columns
Create an AnnData object.
from tiledbsoma import AxisQuery
with census_artifact.open() as store:
experiment = store["census_data"][human]
adata = experiment.axis_query(
"RNA", obs_query=AxisQuery(value_filter=value_filter)
).to_anndata(
X_name="raw",
column_names={
"obs": [
features.assay,
features.cell_type,
features.tissue,
features.disease,
features.suspension_type,
]
},
)
adata.var = adata.var.set_index("feature_id")
adata
Show code cell output
/opt/hostedtoolcache/Python/3.12.13/x64/lib/python3.12/functools.py:912: ImplicitModificationWarning: Transforming to str index.
return dispatch(args[0].__class__)(*args, **kw)
/opt/hostedtoolcache/Python/3.12.13/x64/lib/python3.12/functools.py:912: ImplicitModificationWarning: Transforming to str index.
return dispatch(args[0].__class__)(*args, **kw)
AnnData object with n_obs × n_vars = 117660 × 61497
obs: 'assay', 'cell_type', 'tissue', 'disease', 'suspension_type'
var: 'soma_joinid', 'feature_name', 'feature_type', 'feature_length', 'nnz', 'n_measured_obs'
layers: None (.X)
Train ML models¶
You can either directly train ML models on very large collections of AnnData-like artifacts or on a single concatenated tiledbsoma-like artifact. For pros & cons of these approaches, see this blog post.
On a collection of arrays¶
mapped() caches AnnData objects on disk and creates a map-style dataset that performs a virtual join of the features of the underlying AnnData objects.
from torch.utils.data import DataLoader
census_collection = db.Collection.get(name="cellxgene-census", version="2025-01-30")
dataset = census_collection.mapped(obs_keys=[features.cell_type], join="outer")
dataloader = DataLoader(dataset, batch_size=128, shuffle=True)
for batch in dataloader:
pass
dataset.close()
For more background, see Train a machine learning model on a collection.
On a concatenated array¶
You can create streaming PyTorch dataloaders from tiledbsoma stores using cellxgene_census package.
import cellxgene_census.experimental.ml as census_ml
store = census_artifact.open()
experiment = store["census_data"][human]
experiment_datapipe = census_ml.ExperimentDataPipe(
experiment,
measurement_name="RNA",
X_name="raw",
obs_query=AxisQuery(value_filter=value_filter),
obs_column_names=[features.cell_type],
batch_size=128,
shuffle=True,
soma_chunk_size=10000,
)
experiment_dataloader = census_ml.experiment_dataloader(experiment_datapipe)
for batch in experiment_dataloader:
pass
store.close()
For more background see this guide.