Cell type annotation and pathway analysis 
 ¶
¶
Please make sure that you run the GO Ontology notebook before this one so that your CellType and Pathway registries are populated.
!lamin connect use-cases-registries
Show code cell output
→ connected lamindb: testuser1/use-cases-registries
• to map a local dev directory, call: lamin settings set dev-dir .
# pip install lamindb celltypist gseapy
import lamindb as ln
import bionty as bt
from lamin_usecases import datasets as ds
import scanpy as sc
import matplotlib.pyplot as plt
import celltypist
import gseapy as gp
Show code cell output
→ connected lamindb: testuser1/use-cases-registries
/opt/hostedtoolcache/Python/3.14.6/x64/lib/python3.14/site-packages/celltypist/classifier.py:11: FutureWarning: `__version__` is deprecated, use `importlib.metadata.version('scanpy')` instead
from scanpy import __version__ as scv
An interferon-beta treated dataset¶
A small peripheral blood mononuclear cell dataset that is split into control and stimulated groups. The stimulated group was treated with interferon beta. Let’s load the dataset and perform some preprocessing:
adata = ds.anndata_seurat_ifnb(preprocess=False, populate_registries=True)
adata
Show code cell output
AnnData object with n_obs × n_vars = 13999 × 9731
obs: 'stim'
var: 'symbol'
sc.pp.normalize_total(adata, target_sum=1e4)
sc.pp.log1p(adata)
sc.pp.highly_variable_genes(adata, n_top_genes=2000)
sc.pp.pca(adata, n_comps=20)
sc.pp.neighbors(adata, n_pcs=10)
sc.tl.umap(adata)
Analysis: Cell type annotation using CellTypist¶
model = celltypist.models.Model.load(model="Immune_All_Low.pkl")
Show code cell output
🔎 No available models. Downloading...
📜 Retrieving model list from server https://celltypist.cog.sanger.ac.uk/models/models.json
📚 Total models in list: 61
📂 Storing models in /home/runner/.celltypist/data/models
💾 Downloading model [1/61]: Immune_All_Low.pkl
💾 Downloading model [2/61]: Immune_All_High.pkl
💾 Downloading model [3/61]: Adult_COVID19_PBMC.pkl
💾 Downloading model [4/61]: Adult_CynomolgusMacaque_Hippocampus.pkl
💾 Downloading model [5/61]: Adult_Human_MTG.pkl
💾 Downloading model [6/61]: Adult_Human_PancreaticIslet.pkl
💾 Downloading model [7/61]: Adult_Human_PrefrontalCortex.pkl
💾 Downloading model [8/61]: Adult_Human_Skin.pkl
💾 Downloading model [9/61]: Adult_Human_Vascular.pkl
💾 Downloading model [10/61]: Adult_Mouse_Gut.pkl
💾 Downloading model [11/61]: Adult_Mouse_OlfactoryBulb.pkl
💾 Downloading model [12/61]: Adult_Pig_Hippocampus.pkl
💾 Downloading model [13/61]: Adult_RhesusMacaque_Hippocampus.pkl
💾 Downloading model [14/61]: Adult_cHSPCs_Illumina.pkl
💾 Downloading model [15/61]: Adult_cHSPCs_Ultima.pkl
💾 Downloading model [16/61]: Autopsy_COVID19_Lung.pkl
💾 Downloading model [17/61]: COVID19_HumanChallenge_Blood.pkl
💾 Downloading model [18/61]: COVID19_Immune_Landscape.pkl
💾 Downloading model [19/61]: Cells_Adult_Breast.pkl
💾 Downloading model [20/61]: Cells_Fetal_Lung.pkl
💾 Downloading model [21/61]: Cells_Human_Tonsil.pkl
💾 Downloading model [22/61]: Cells_Intestinal_Tract.pkl
💾 Downloading model [23/61]: Cells_Lung_Airway.pkl
💾 Downloading model [24/61]: Developing_Human_Brain.pkl
💾 Downloading model [25/61]: Developing_Human_Gonads.pkl
💾 Downloading model [26/61]: Developing_Human_Hippocampus.pkl
💾 Downloading model [27/61]: Developing_Human_Organs.pkl
💾 Downloading model [28/61]: Developing_Human_Thymus.pkl
💾 Downloading model [29/61]: Developing_Mouse_Brain.pkl
💾 Downloading model [30/61]: Developing_Mouse_Hippocampus.pkl
💾 Downloading model [31/61]: Fetal_Human_AdrenalGlands.pkl
💾 Downloading model [32/61]: Fetal_Human_Pancreas.pkl
💾 Downloading model [33/61]: Fetal_Human_Pituitary.pkl
💾 Downloading model [34/61]: Fetal_Human_Retina.pkl
💾 Downloading model [35/61]: Fetal_Human_Skin.pkl
💾 Downloading model [36/61]: Healthy_Adult_Heart.pkl
💾 Downloading model [37/61]: Healthy_COVID19_PBMC.pkl
💾 Downloading model [38/61]: Healthy_Human_Liver.pkl
💾 Downloading model [39/61]: Healthy_Mouse_Liver.pkl
💾 Downloading model [40/61]: Human_AdultAged_Hippocampus.pkl
💾 Downloading model [41/61]: Human_Colorectal_Cancer.pkl
💾 Downloading model [42/61]: Human_Developmental_Retina.pkl
💾 Downloading model [43/61]: Human_Embryonic_YolkSac.pkl
💾 Downloading model [44/61]: Human_Endometrium_Atlas.pkl
💾 Downloading model [45/61]: Human_IPF_Lung.pkl
💾 Downloading model [46/61]: Human_Longitudinal_Hippocampus.pkl
💾 Downloading model [47/61]: Human_Lung_Atlas.pkl
💾 Downloading model [48/61]: Human_PF_Lung.pkl
💾 Downloading model [49/61]: Human_Placenta_Decidua.pkl
💾 Downloading model [50/61]: Lethal_COVID19_Lung.pkl
💾 Downloading model [51/61]: Mouse_Dendritic_Subtypes.pkl
💾 Downloading model [52/61]: Mouse_Dentate_Gyrus.pkl
💾 Downloading model [53/61]: Mouse_Isocortex_Hippocampus.pkl
💾 Downloading model [54/61]: Mouse_Postnatal_DentateGyrus.pkl
💾 Downloading model [55/61]: Mouse_Whole_Brain.pkl
💾 Downloading model [56/61]: Nuclei_Human_InnerEar.pkl
💾 Downloading model [57/61]: Nuclei_Lung_Airway.pkl
💾 Downloading model [58/61]: PaediatricAdult_COVID19_Airway.pkl
💾 Downloading model [59/61]: PaediatricAdult_COVID19_PBMC.pkl
💾 Downloading model [60/61]: Pan_Fetal_Human.pkl
💾 Downloading model [61/61]: Thymus_Allograft_PBMC.pkl
predictions = celltypist.annotate(
adata, model="Immune_All_Low.pkl", majority_voting=True
)
adata.obs["cell_type_celltypist"] = predictions.predicted_labels.majority_voting
Show code cell output
🔬 Input data has 13999 cells and 9731 genes
🔗 Matching reference genes in the model
🧬 3641 features used for prediction
⚖️ Scaling input data
🖋️ Predicting labels
✅ Prediction done!
👀 Detected a neighborhood graph in the input object, will run over-clustering on the basis of it
⛓️ Over-clustering input data with resolution set to 10
🗳️ Majority voting the predictions
✅ Majority voting done!
adata.obs["cell_type_celltypist"] = bt.CellType.standardize(
adata.obs["cell_type_celltypist"]
)
sc.pl.umap(
adata,
color=["cell_type_celltypist", "stim"],
frameon=False,
legend_fontsize=10,
wspace=0.4,
)
Show code cell output
... storing 'cell_type_celltypist' as categorical
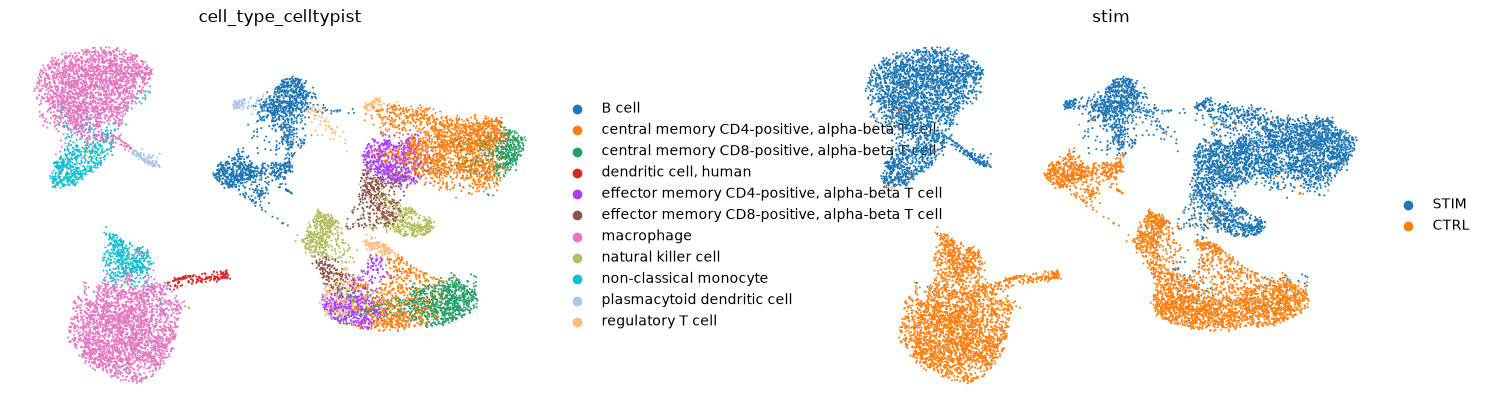
Analysis: Pathway enrichment analysis using Enrichr¶
This analysis is based on the GSEApy scRNA-seq Example.
First, we compute differentially expressed genes using a Wilcoxon test between stimulated and control cells.
# compute differentially expressed genes
sc.tl.rank_genes_groups(
adata,
groupby="stim",
use_raw=False,
method="wilcoxon",
groups=["STIM"],
reference="CTRL",
)
rank_genes_groups_df = sc.get.rank_genes_groups_df(adata, "STIM")
rank_genes_groups_df.head()
Show code cell output
| names | scores | logfoldchanges | pvals | pvals_adj | |
|---|---|---|---|---|---|
| 0 | ISG15 | 99.317345 | 7.148426 | 0.0 | 0.0 |
| 1 | ISG20 | 96.655273 | 5.100947 | 0.0 | 0.0 |
| 2 | IFI6 | 94.771217 | 5.828259 | 0.0 | 0.0 |
| 3 | IFIT3 | 92.449509 | 7.450851 | 0.0 | 0.0 |
| 4 | IFIT1 | 90.678734 | 8.078267 | 0.0 | 0.0 |
Next, we filter out up/down-regulated differentially expressed gene sets:
degs_up = rank_genes_groups_df[
(rank_genes_groups_df["logfoldchanges"] > 0)
& (rank_genes_groups_df["pvals_adj"] < 0.05)
]
degs_dw = rank_genes_groups_df[
(rank_genes_groups_df["logfoldchanges"] < 0)
& (rank_genes_groups_df["pvals_adj"] < 0.05)
]
degs_up.shape, degs_dw.shape
Show code cell output
((535, 5), (939, 5))
Run pathway enrichment analysis on DEGs and plot top 10 pathways:
enr_up = gp.enrichr(degs_up.names, gene_sets="GO_Biological_Process_2023").res2d
gp.dotplot(enr_up, figsize=(2, 3), title="Up", cmap=plt.cm.autumn_r);
Show code cell output

enr_dw = gp.enrichr(degs_dw.names, gene_sets="GO_Biological_Process_2023").res2d
gp.dotplot(enr_dw, figsize=(2, 3), title="Down", cmap=plt.cm.winter_r);
Show code cell output

Annotate & save dataset¶
gRegister new features and labels (check out more details here):
ln.Feature(name="cell_type_celltypist", dtype=bt.CellType).save()
ln.Feature(name="stim", dtype=str).save()
obs_schema = ln.Schema(
name="celltype_obs_schema",
features=[
ln.Feature(name="cell_type_celltypist", dtype=bt.CellType).save(),
ln.Feature(name="stim", dtype=str).save(),
],
).save()
var_schema = ln.Schema(
name="gene_var_schema",
itype=bt.Gene,
).save()
schema = ln.Schema(
name="anndata_seurat_ifnb_schema",
slots={"obs": obs_schema, "var.T": var_schema},
otype="AnnData",
).save()
Show code cell output
→ returning feature with same name: 'cell_type_celltypist'
→ returning feature with same name: 'stim'
Register dataset using an Artifact object:
artifact = ln.Artifact.from_anndata(
adata,
description="seurat_ifnb_activated_Bcells",
schema=schema,
).save()
artifact.describe()
Show code cell output
! no run & transform got linked, call `ln.track()` & re-run
→ loading artifact into memory for validation
→ returning schema with same hash: Schema(uid='jjbK3qe9JCRNVruc', is_type=False, name='celltype_obs_schema', description=None, n_members=2, coerce=None, flexible=False, itype='Feature', otype=None, suffix=None, hash='wzQGVwDU6KjzQbEFxzDwAg', minimal_set=True, ordered_set=False, maximal_set=False, branch_id=1, created_on_id=1, space_id=1, created_by_id=1, run_id=None, type_id=None, created_at=2026-08-03 16:57:13 UTC, is_locked=False)
→ not annotating with 11053 features for slot var.T as it exceeds 1000 (ln.settings.annotation.n_max_records)
Artifact: (0000) | description: seurat_ifnb_activated_Bcells ├── uid: FNzIlmsw6p5GXzvy0000 kind: dataset │ otype: AnnData hash: dh69gOJjdwMGjB-u-hVhGj │ size: 202.0 MB branch: main │ space: all created_at: 2026-08-03 16:57:15 UTC │ created_by: testuser1 n_observations: 13999 │ schema: anndata_seurat_ifnb_schema ├── storage/path: │ /home/runner/work/lamin-usecases/lamin-usecases/docs/use-cases-registries/.lamindb/FNzIlmsw6p5GXzvy0000.h5ad ├── Dataset features │ ├── obs (2) │ │ cell_type_celltypist bionty.CellType B cell, central memory CD4-positive, a… │ │ stim str │ └── var.T (11053 bionty.Gene.sym… └── Labels └── .cell_types bionty.CellType B cell, dendritic cell, human, natural…
Manage pathway objects¶
Let’s create two schemas (two feature sets) for degs_up and degs_dw:
schema_degs_up = ln.Schema.from_values(
degs_up.names,
bt.Gene.symbol,
name="Up-regulated DEGs STIM vs CTRL",
organism="human",
).save()
schema_degs_dw = ln.Schema.from_values(
degs_dw.names,
bt.Gene.symbol,
name="Down-regulated DEGs STIM vs CTRL",
organism="human",
).save()
Link the top 10 pathways to the corresponding differentially expressed genes:
def parse_ontology_id_from_enrichr_results(key):
"""Parse out the ontology id.
"ATF6-mediated Unfolded Protein Response (GO:0036500)" -> ("GO:0036500", "ATF6-mediated Unfolded Protein Response")
"""
id = key.split(" ")[-1].replace("(", "").replace(")", "")
name = key.replace(f" ({id})", "")
return (id, name)
# get ontology ids for the top 10 pathways
enr_up_top10 = [
pw_id[0]
for pw_id in enr_up.head(10).Term.apply(parse_ontology_id_from_enrichr_results)
]
enr_dw_top10 = [
pw_id[0]
for pw_id in enr_dw.head(10).Term.apply(parse_ontology_id_from_enrichr_results)
]
# get pathway objects
enr_up_top10_pathways = bt.Pathway.from_values(enr_up_top10, bt.Pathway.ontology_id)
enr_dw_top10_pathways = bt.Pathway.from_values(enr_dw_top10, bt.Pathway.ontology_id)
Associate the pathways to the differentially expressed genes:
schema_degs_up.pathways.set(enr_up_top10_pathways)
schema_degs_dw.pathways.set(enr_dw_top10_pathways)
schema_degs_up.pathways.to_list("name")
Show code cell output
['cellular response to cytokine stimulus',
'defense response to symbiont',
'defense response to virus',
'negative regulation of viral genome replication',
'negative regulation of viral process',
'positive regulation of cytokine production',
'regulation of viral genome replication',
'response to cytokine',
'response to interferon-beta',
'response to type II interferon']
With this, we stored the result of the differential expression analysis via schema objects where each schema object links a gene set and its set of enriched pathways in the dataset.
This allows queries along the lines below.
Query pathways¶
Querying for pathways contains “interferon-beta” in the name:
bt.Pathway.filter(name__contains="interferon-beta").to_dataframe()
Show code cell output
| uid | name | ontology_id | abbr | synonyms | description | is_locked | created_at | branch_id | created_on_id | space_id | created_by_id | run_id | source_id | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| id | ||||||||||||||
| 4885 | 3VZq4dMeKlygVU | response to interferon-beta | GO:0035456 | None | response to fibroblast interferon|response to ... | Any Process That Results In A Change In State ... | False | 2026-08-03 16:52:57.483000+00:00 | 1 | 1 | 1 | 1 | None | 35 |
| 4277 | 54R2a0elVmUTQY | regulation of interferon-beta production | GO:0032648 | None | regulation of IFN-beta production | Any Process That Modulates The Frequency, Rate... | False | 2026-08-03 16:52:57.428000+00:00 | 1 | 1 | 1 | 1 | None | 35 |
| 3121 | 3x0xmK1yUYa5Uk | positive regulation of interferon-beta production | GO:0032728 | None | up-regulation of interferon-beta production|po... | Any Process That Activates Or Increases The Fr... | False | 2026-08-03 16:52:57.330000+00:00 | 1 | 1 | 1 | 1 | None | 35 |
| 2150 | 1NzHDJDiVT0xDa | negative regulation of interferon-beta production | GO:0032688 | None | downregulation of interferon-beta production|n... | Any Process That Stops, Prevents, Or Reduces T... | False | 2026-08-03 16:52:57.062000+00:00 | 1 | 1 | 1 | 1 | None | 35 |
| 689 | 1l4z0v8WGwcxuN | cellular response to interferon-beta | GO:0035458 | None | cellular response to fiblaferon|cellular respo... | Any Process That Results In A Change In State ... | False | 2026-08-03 16:52:56.926000+00:00 | 1 | 1 | 1 | 1 | None | 35 |
Query pathways from a gene:
bt.Pathway.filter(genes__symbol="IFITM1").to_dataframe()
Show code cell output
! truncated query result to limit=20 Pathway objects
| uid | name | ontology_id | abbr | synonyms | description | is_locked | created_at | branch_id | created_on_id | space_id | created_by_id | run_id | source_id | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| id | ||||||||||||||
| 5251 | 3dRO41YW68H1ME | type I interferon-mediated signaling pathway | GO:0060337 | None | type I interferon-activated signaling pathway|... | The Series Of Molecular Signals Initiated By T... | False | 2026-08-03 16:52:57.516000+00:00 | 1 | 1 | 1 | 1 | None | 35 |
| 4924 | 7m7ayBKAWfxEoI | response to type II interferon | GO:0034341 | None | response to immune interferon | Any Process That Results In A Change In State ... | False | 2026-08-03 16:52:57.488000+00:00 | 1 | 1 | 1 | 1 | None | 35 |
| 4885 | 3VZq4dMeKlygVU | response to interferon-beta | GO:0035456 | None | response to fibroblast interferon|response to ... | Any Process That Results In A Change In State ... | False | 2026-08-03 16:52:57.483000+00:00 | 1 | 1 | 1 | 1 | None | 35 |
| 4884 | 1Mkdeon3xT6DKg | response to interferon-alpha | GO:0035455 | None | response to lymphoblastoid interferon|response... | Any Process That Results In A Change In State ... | False | 2026-08-03 16:52:57.483000+00:00 | 1 | 1 | 1 | 1 | None | 35 |
| 4859 | 6WvSjRrrPb7dFT | response to cytokine | GO:0034097 | None | response to cytokine stimulus | Any Process That Results In A Change In State ... | False | 2026-08-03 16:52:57.483000+00:00 | 1 | 1 | 1 | 1 | None | 35 |
| 4792 | 41BNWUg3sxpyoS | regulation of viral genome replication | GO:0045069 | None | None | Any Process That Modulates The Frequency, Rate... | False | 2026-08-03 16:52:57.477000+00:00 | 1 | 1 | 1 | 1 | None | 35 |
| 4498 | 2qCl1QmE5rYFHy | regulation of osteoblast differentiation | GO:0045667 | None | None | Any Process That Modulates The Frequency, Rate... | False | 2026-08-03 16:52:57.450000+00:00 | 1 | 1 | 1 | 1 | None | 35 |
| 4050 | 1co8167Esy6IID | regulation of cell population proliferation | GO:0042127 | None | None | Any Process That Modulates The Frequency, Rate... | False | 2026-08-03 16:52:57.412000+00:00 | 1 | 1 | 1 | 1 | None | 35 |
| 4046 | 4LqrUcXAKER15Q | regulation of cell migration | GO:0030334 | None | None | Any Process That Modulates The Frequency, Rate... | False | 2026-08-03 16:52:57.412000+00:00 | 1 | 1 | 1 | 1 | None | 35 |
| 3300 | 66TxxRHcFq47cP | positive regulation of osteoblast differentiation | GO:0045669 | None | up-regulation of osteoblast differentiation|up... | Any Process That Activates Or Increases The Fr... | False | 2026-08-03 16:52:57.346000+00:00 | 1 | 1 | 1 | 1 | None | 35 |
| 2945 | 2xtOQMxdMWpZOC | positive regulation of cell differentiation | GO:0045597 | None | up-regulation of cell differentiation|up regul... | Any Process That Activates Or Increases The Fr... | False | 2026-08-03 16:52:57.134000+00:00 | 1 | 1 | 1 | 1 | None | 35 |
| 2442 | 2DyRG9whXU3xJB | negative regulation of viral process | GO:0048525 | None | None | Any Process That Stops, Prevents, Or Reduces T... | False | 2026-08-03 16:52:57.090000+00:00 | 1 | 1 | 1 | 1 | None | 35 |
| 2440 | KduBJgyedVzK2X | negative regulation of viral genome replication | GO:0045071 | None | down-regulation of viral genome replication|do... | Any Process That Stops, Prevents, Or Reduces T... | False | 2026-08-03 16:52:57.090000+00:00 | 1 | 1 | 1 | 1 | None | 35 |
| 2034 | 3rjhK2RNOFztVV | negative regulation of cellular process | GO:0048523 | None | downregulation of cellular process|down-regula... | Any Process That Stops, Prevents, Or Reduces T... | False | 2026-08-03 16:52:57.051000+00:00 | 1 | 1 | 1 | 1 | None | 35 |
| 2024 | 701eu1FQnCjB3N | negative regulation of cell population prolife... | GO:0008285 | None | down-regulation of cell proliferation|downregu... | Any Process That Stops, Prevents Or Reduces Th... | False | 2026-08-03 16:52:57.051000+00:00 | 1 | 1 | 1 | 1 | None | 35 |
| 2023 | 51PBemXLpN96fT | negative regulation of cell motility | GO:2000146 | None | negative regulation of cell locomotion|negativ... | Any Process That Stops, Prevents, Or Reduces T... | False | 2026-08-03 16:52:57.051000+00:00 | 1 | 1 | 1 | 1 | None | 35 |
| 2021 | 5dL3FOu3nuaoMl | negative regulation of cell migration | GO:0030336 | None | downregulation of cell migration|down regulati... | Any Process That Stops, Prevents, Or Reduces T... | False | 2026-08-03 16:52:57.051000+00:00 | 1 | 1 | 1 | 1 | None | 35 |
| 1402 | 2QMDbGJZh3R1bF | interferon-mediated signaling pathway | GO:0140888 | None | interferon signaling pathway|interferon-activa... | The Series Of Molecular Signals Initiated By T... | False | 2026-08-03 16:52:56.983000+00:00 | 1 | 1 | 1 | 1 | None | 35 |
| 1344 | 36S4bCrBw6ebbo | host-mediated suppression of symbiont invasion | GO:0046597 | None | negative regulation of viral penetration into ... | A Process In Which A Host Inhibits Or Disrupts... | False | 2026-08-03 16:52:56.978000+00:00 | 1 | 1 | 1 | 1 | None | 35 |
| 880 | 43JHYr10qy8F6o | defense response to virus | GO:0051607 | None | defense response to viruses|antiviral response... | Reactions Triggered In Response To The Presenc... | False | 2026-08-03 16:52:56.942000+00:00 | 1 | 1 | 1 | 1 | None | 35 |
Query artifacts from a pathway:
ln.Artifact.filter(feature_sets__pathways__name__icontains="interferon-beta").first()
Query schemas from a pathway to learn from which geneset this pathway was computed:
pathway = bt.Pathway.get(ontology_id="GO:0035456")
pathway
Show code cell output
Pathway(uid='3VZq4dMeKlygVU', name='response to interferon-beta', ontology_id='GO:0035456', abbr=None, synonyms='response to fibroblast interferon|response to interferon beta|response to fiblaferon', description='Any Process That Results In A Change In State Or Activity Of A Cell Or An Organism (In Terms Of Movement, Secretion, Enzyme Production, Gene Expression, Etc.) As A Result Of An Interferon-Beta Stimulus. Interferon-Beta Is A Type I Interferon.', branch_id=1, created_on_id=1, space_id=1, created_by_id=1, run_id=None, source_id=35, created_at=2026-08-03 16:52:57 UTC, is_locked=False)
degs = ln.Schema.get(pathways__ontology_id=pathway.ontology_id)
Get the list of genes that are differentially expressed and belong to this pathway:
contributing_genes = pathway.genes.all() & degs.genes.all()
contributing_genes.to_list("symbol")
Show code cell output
['MNDA',
'AIM2',
'PNPT1',
'PLSCR1',
'OAS1',
'IRF1',
'CALM1',
'STAT1',
'IFI16',
'IFITM2',
'IFITM1',
'IFITM3',
'BST2',
'SHFL',
'XAF1']
# clean up test instance
!rm -r ./use-cases-registries
!lamin delete --force use-cases-registries