Flow cytometry 
 ¶
¶
You’ll learn how to manage a growing number of flow cytometry datasets as a single queryable collection.
Specifically, you will
read a single
.fcsfile as anAnnDataand seed a versioned collection with it (, current page)
append a new dataset (a new
.fcsfile) to create a new version of the collection ()
query individual files and cell markers (
)
analyze the collection and store results as plots (
)
# !pip install 'lamindb[jupyter,bionty]'
!lamin init --storage ./test-facs --modules bionty
Show code cell output
→ initialized lamindb: testuser1/test-facs
import lamindb as ln
import bionty as bt
import readfcs
bt.settings.organism = "human" # globally set organism to human
ln.track("OWuTtS4SApon0000")
Show code cell output
→ connected lamindb: testuser1/test-facs
→ created Transform('OWuTtS4SApon0000', key='facs.ipynb'), started new Run('9YSlMmuPPZKdZrgj') at 2026-07-30 06:13:10 UTC
→ notebook imports: bionty==2.4.2 lamindb-core==2.8.1 pytometry==0.1.6 readfcs==2.1.0 scanpy==1.12.3
Ingest a first artifact¶
Access  ¶
¶
We start with a flow cytometry file from Alpert et al., Nat. Med. (2019).
Calling the following function downloads the artifact and pre-populates a few relevant registries:
ln.core.datasets.file_fcs_alpert19(populate_registries=True)
Show code cell output
/opt/hostedtoolcache/Python/3.12.13/x64/lib/python3.12/functools.py:912: ImplicitModificationWarning: Transforming to str index.
return dispatch(args[0].__class__)(*args, **kw)
PosixPath('Alpert19.fcs')
We use readfcs to read the raw fcs file into memory and create an AnnData object:
adata = readfcs.read("Alpert19.fcs")
adata
Show code cell output
/opt/hostedtoolcache/Python/3.12.13/x64/lib/python3.12/functools.py:912: ImplicitModificationWarning: Transforming to str index.
return dispatch(args[0].__class__)(*args, **kw)
AnnData object with n_obs × n_vars = 166537 × 40
var: 'n', 'channel', 'marker', 'PnB', 'PnE', 'PnR'
uns: 'meta'
It has the following features:
adata.var.head(10)
Show code cell output
| n | channel | marker | PnB | PnE | PnR | |
|---|---|---|---|---|---|---|
| Time | 1 | Time | 32 | 0,0 | 2097152 | |
| Cell_length | 2 | Cell_length | 32 | 0,0 | 128 | |
| CD57 | 3 | (In113)Dd | CD57 | 32 | 0,0 | 8192 |
| Dead | 4 | (In115)Dd | Dead | 32 | 0,0 | 4096 |
| (Ba138)Dd | 5 | (Ba138)Dd | 32 | 0,0 | 4096 | |
| Bead | 6 | (Ce140)Dd | Bead | 32 | 0,0 | 16384 |
| CD19 | 7 | (Nd142)Dd | CD19 | 32 | 0,0 | 4096 |
| CD4 | 8 | (Nd143)Dd | CD4 | 32 | 0,0 | 4096 |
| CD8 | 9 | (Nd144)Dd | CD8 | 32 | 0,0 | 4096 |
| IgD | 10 | (Nd146)Dd | IgD | 32 | 0,0 | 8192 |
Transform: normalize  ¶
¶
In this use case, we’d like to ingest & store curated data, and hence, we split signal and normalize using the pytometry package.
import pytometry as pm
First, we’ll split the signal from heigh and area metadata:
pm.pp.split_signal(adata, var_key="channel", data_type="cytof")
Show code cell output
'area' is not in adata.var['signal_type']. Return all.
/opt/hostedtoolcache/Python/3.12.13/x64/lib/python3.12/site-packages/pytometry/pp/_process_data.py:231: FutureWarning: Use var (e.g. `k in adata.var` or `str(adata.var.columns.tolist())`) instead of AnnData.var_keys, AnnData.var_keys is deprecated and will be removed in the future.
if key_in not in adata.var_keys():
/opt/hostedtoolcache/Python/3.12.13/x64/lib/python3.12/site-packages/pytometry/pp/_process_data.py:65: FutureWarning: Use var (e.g. `k in adata.var` or `str(adata.var.columns.tolist())`) instead of AnnData.var_keys, AnnData.var_keys is deprecated and will be removed in the future.
elif var_key in adata.var_keys():
adata
Show code cell output
AnnData object with n_obs × n_vars = 166537 × 40
var: 'n', 'channel', 'marker', 'PnB', 'PnE', 'PnR', 'signal_type'
uns: 'meta'
Normalize the collection:
pm.tl.normalize_arcsinh(adata, cofactor=150)
Note
If the collection was a flow collection, you’ll also have to compensate the data, if possible. The metadata should contain a compensation matrix, which could then be run by the pytometry compensation function. In the case here, its a cyTOF collection, which doesn’t (really) require compensation.
Validate: cell markers  ¶
¶
First, we validate features in .var using CellMarker:
validated = bt.CellMarker.validate(adata.var.index)
Show code cell output
! 13 unique terms (32.50%) are not validated for name: 'Time', 'Cell_length', 'Dead', '(Ba138)Dd', 'Bead', 'CD19', 'CD4', 'IgD', 'CD11b', 'CD14', ...
We see that many features aren’t validated because they’re not standardized.
Hence, let’s standardize feature names & validate again:
adata.var.index = bt.CellMarker.standardize(adata.var.index)
validated = bt.CellMarker.validate(adata.var.index)
Show code cell output
! 5 unique terms (12.50%) are not validated for name: 'Time', 'Cell_length', 'Dead', '(Ba138)Dd', 'Bead'
The remaining non-validated features don’t appear to be cell markers but rather metadata features.
Let’s move them into adata.obs:
adata.obs = adata[:, ~validated].to_df()
adata = adata[:, validated].copy()
Now we have a clean panel of 35 validated cell markers:
validated = bt.CellMarker.validate(adata.var.index)
assert all(validated) # all markers are validated
Register: metadata  ¶
¶
Next, let’s register the metadata features we moved to .obs.
For this, we create one feature record for each column in the .obs dataframe:
features = ln.Feature.from_dataframe(adata.obs)
ln.save(features)
We use the Experimental Factor Ontology through Bionty to create a “FACS” label:
bt.ExperimentalFactor.public().search("FACS").head(2) # search the public ontology
Show code cell output
| name | definition | synonyms | parents | |
|---|---|---|---|---|
| ontology_id | ||||
| EFO:0009108 | fluorescence-activated cell sorting | A Flow Cytometry Assay That Provides A Method ... | FAC sorting|FACS | [] |
| EFO:0700083 | fetal anticonvulsant syndrome | Syndromes Associated With Congenital Malformat... | fetal AEDS|foetal antiepileptic drug syndrome|... | [] |
We found one for “FACS”, let’s save it to our in-house registry:
# import the FACS record from the public ontology and save it to the registry
facs = bt.ExperimentalFactor.from_source(ontology_id="EFO:0009108")
facs.save()
Show code cell output
ExperimentalFactor(uid='36GhLFoEqEva7C', name='fluorescence-activated cell sorting', ontology_id='EFO:0009108', abbr=None, synonyms='FAC sorting|FACS', description='A Flow Cytometry Assay That Provides A Method For Sorting A Heterogeneous Mixture Of Biological Cells Into Two Or More Containers, One Cell At A Time, Based Upon The Specific Light Scattering And Fluorescent Characteristics Of Each Cell.
The Cells Are Suspended In A Stream Of Fluid And Forced Individually Through A Vibrating Nozzle, Then Exposed To A Laser Beam And The Resulting Fluorescence And Scattered Light Is Detected. Finally The Cells Are Sorted By Applying An Electrical Charge To Droplets Of The Fluid And Deflecting It To The Left Or Right Using Charged Electrodes.', branch_id=1, created_on_id=1, space_id=1, created_by_id=1, run_id=1, source_id=31, created_at=2026-07-30 06:13:16 UTC, is_locked=False)
We don’t find one for “CyToF”, however, so, let’s create it without importing from a public ontology but label it as a child of “is_cytometry_assay”:
cytof = bt.ExperimentalFactor(name="CyTOF")
cytof.save()
is_cytometry_assay = bt.ExperimentalFactor(name="is_cytometry_assay")
is_cytometry_assay.save()
cytof.parents.add(is_cytometry_assay)
facs.parents.add(is_cytometry_assay)
is_cytometry_assay.view_parents(with_children=True)
Show code cell output

Let us look at the content of the registry:
bt.ExperimentalFactor.to_dataframe()
Show code cell output
| uid | name | ontology_id | abbr | synonyms | description | is_locked | created_at | branch_id | created_on_id | space_id | created_by_id | run_id | source_id | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| id | ||||||||||||||
| 3 | 21Qymj4Q8aWo2j | is_cytometry_assay | None | None | None | None | False | 2026-07-30 06:13:16.419000+00:00 | 1 | 1 | 1 | 1 | 1 | NaN |
| 2 | ogoPdeOkdBFBJm | CyTOF | None | None | None | None | False | 2026-07-30 06:13:16.415000+00:00 | 1 | 1 | 1 | 1 | 1 | NaN |
| 1 | 36GhLFoEqEva7C | fluorescence-activated cell sorting | EFO:0009108 | None | FAC sorting|FACS | A Flow Cytometry Assay That Provides A Method ... | False | 2026-07-30 06:13:16.047000+00:00 | 1 | 1 | 1 | 1 | 1 | 31.0 |
Register: save & annotate with metadata ¶
var_schema = ln.Schema(
name="FACS-cell-markers",
itype=bt.CellMarker,
).save()
obs_schema = ln.Schema(
name="FACS-sample-metadata",
itype=ln.Feature,
flexible=True,
).save()
schema = ln.Schema(
name="FACS-AnnData-schema",
otype="AnnData",
slots={"obs": obs_schema, "var.T": var_schema},
).save()
curator = ln.curators.AnnDataCurator(adata, schema=schema)
artifact = curator.save_artifact(description="Alpert19")
Add more labels:
experimental_factors = bt.ExperimentalFactor.lookup()
organisms = bt.Organism.lookup()
artifact.labels.add(experimental_factors.cytof)
artifact.labels.add(organisms.human)
Inspect the saved artifact¶
Inspect features on a high level:
artifact.features
Show code cell output
Artifact: (0000) | description: Alpert19 └── Dataset features ├── obs (5) │ (Ba138)Dd float │ Bead float │ Cell_length float │ Dead float │ Time float └── var.T (35 bionty.CellMarker) CD11B num CD11c num CD16 num CD27 num CD3 num CD38 num CD45RA num CD57 num CD8 num CD85j num CD86 num CD94 num CXCR3 num CXCR5 num Ccr6 num Ccr7 num Cd14 num Cd19 num Cd4 num Igd num
Inspect low-level features in .var:
artifact.features.slots["var.T"].members.to_dataframe().head()
Show code cell output
! truncated query result to limit=20 CellMarker objects
| uid | abbr | synonyms | description | name | gene_symbol | ncbi_gene_id | uniprotkb_id | is_locked | created_at | branch_id | created_on_id | space_id | created_by_id | run_id | source_id | organism_id | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| id | |||||||||||||||||
| 1 | 5R8E1YHbOROIdm | None | None | CD57 | B3GAT1 | 27087 | Q9P2W7 | False | 2026-07-30 06:13:13.436000+00:00 | 1 | 1 | 1 | 1 | 1 | 21 | 1 | |
| 2 | 19Sxm5VN87z8Zo | None | None | Cd19 | CD19 | 930 | P15391 | False | 2026-07-30 06:13:13.436000+00:00 | 1 | 1 | 1 | 1 | 1 | 21 | 1 | |
| 3 | 5CbKd6B4ILaqJQ | None | None | Cd4 | CD4 | 920 | B4DT49 | False | 2026-07-30 06:13:13.436000+00:00 | 1 | 1 | 1 | 1 | 1 | 21 | 1 | |
| 4 | 1xRpnOHIkdyEXB | None | None | CD8 | CD8A | 925 | P01732 | False | 2026-07-30 06:13:13.436000+00:00 | 1 | 1 | 1 | 1 | 1 | 21 | 1 | |
| 5 | 7fdKraUfUF8wQ5 | None | None | Igd | None | None | None | False | 2026-07-30 06:13:13.436000+00:00 | 1 | 1 | 1 | 1 | 1 | 21 | 1 |
Use auto-complete for marker names in the var featureset:
markers = artifact.features.slots["var.T"].members.lookup()
markers.cd14
Show code cell output
CellMarker(uid='3x83PW1Qiafddi', abbr=None, synonyms='', description=None, name='Cd14', gene_symbol='CD14', ncbi_gene_id='4695', uniprotkb_id='O43678', branch_id=1, created_on_id=1, space_id=1, created_by_id=1, run_id=1, source_id=21, organism_id=1, created_at=2026-07-30 06:13:13 UTC, is_locked=False)
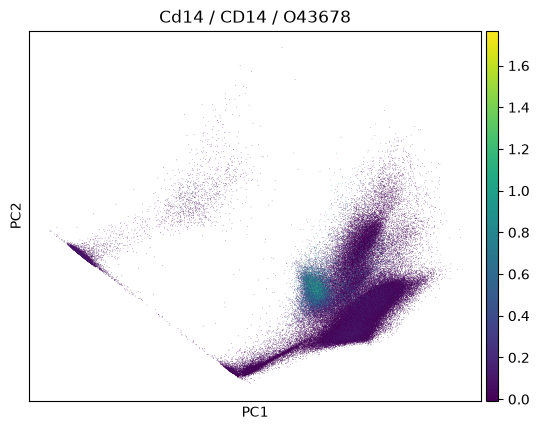
In a plot, we can now easily also show gene symbol and Uniprot ID:
import scanpy as sc
sc.pp.pca(adata)
sc.pl.pca(
adata,
color=markers.cd14.name,
title=(
f"{markers.cd14.name} / {markers.cd14.gene_symbol} /"
f" {markers.cd14.uniprotkb_id}"
),
)
Show code cell output
Create a collection from the artifact¶
ln.Collection(artifact, key="My versioned cytometry collection", version="1").save()
Show code cell output
Collection(uid='vvMAPTmaf0Kamy8V0000', key='My versioned cytometry collection', description=None, hash='gKJFJJqKr0KQ6Q6EaoGrSA', reference=None, reference_type=None, meta_artifact=None, branch_id=1, created_on_id=1, space_id=1, created_by_id=1, run_id=1, schema_id=None, created_at=2026-07-30 06:13:18 UTC, is_locked=False, version_tag='1', is_latest=True)