Spatial RNA-seq 
 ¶
¶
Here, you’ll learn how to manage spatial datasets:
query & analyze spatial datasets (
)
create and share interactive visualizations with vitessce (
)
curate and ingest spatial data (
)
load the collection into memory & train a ML model (
)
Spatial omics data integrates molecular profiling (e.g., transcriptomics, proteomics) with spatial information, preserving the spatial organization of cells and tissues. It enables high-resolution mapping of molecular activity within biological contexts, crucial for understanding cellular interactions and microenvironments.
Many different spatial technologies such as multiplexed imaging, spatial transcriptomics, spatial proteomics, whole slide imaging, spatial metabolomics, and 3D tissue reconstruction exist which can all be stored in the SpatialData data framework. For more details we refer to the original publication:
Marconato, L., Palla, G., Yamauchi, K.A. et al. SpatialData: an open and universal data framework for spatial omics. Nat Methods 22, 58–62 (2025). https://doi.org/10.1038/s41592-024-02212-x
Note
A collection of curated spatial datasets in SpatialData format is available on the scverse/spatialdata-db instance.
spatial data vs SpatialData terminology
When we mention spatial data, we refer to data from spatial assays, such as spatial transcriptomics or proteomics, that includes spatial coordinates to represent the organization of molecular features in tissue. When we refer SpatialData, we mean spatial omics data stored in the scverse SpatialData framework.
Query and analyze spatial data¶
# pip install lamindb spatialdata spatialdata-plot squidpy
!lamin init --storage ./test-spatial --modules bionty
Show code cell output
→ initialized lamindb: testuser1/test-spatial
import warnings
warnings.filterwarnings("ignore")
import lamindb as ln
import scanpy as sc
import spatialdata as sd
import spatialdata_plot as pl
import squidpy as sq
from matplotlib import pyplot as plt
ln.track()
Show code cell output
→ connected lamindb: testuser1/test-spatial
→ created Transform('Zt3BXoG4r47J0000', key='spatial.ipynb'), started new Run('8CIGOdTiMKm9uptO') at 2026-08-03 16:50:23 UTC
→ notebook imports: lamindb-core==2.9.0 matplotlib==3.11.1 scanpy==1.12.3 spatialdata-plot==0.4.0 spatialdata==0.8.0 squidpy==1.8.3
• tip: to identify the notebook across renames, pass the uid: ln.track("Zt3BXoG4r47J")
Query by biological metadata¶
We’ll work with a human lung cancer dataset generated using 10x Genomics Xenium platform and available in a public instance. This FFPE (formalin-fixed paraffin-embedded) tissue sample includes spatial gene expression profiles.
Create the central query object of our public lamindata instance:
db = ln.DB("laminlabs/lamindata")
Show code cell output
! database has module pertdb, configure it: lamin settings modules set bionty,pertdb
Now query the database to find Xenium datasets from lung tissue:
all_xenium_data = db.Artifact.filter(
assay="Xenium Spatial Gene Expression", tissue="lung"
)
all_xenium_data.to_dataframe()
Show code cell output
| uid | key | description | suffix | kind | otype | size | hash | n_files | n_observations | ... | is_latest | is_locked | created_at | branch_id | created_on_id | space_id | storage_id | run_id | schema_id | created_by_id | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| id | |||||||||||||||||||||
| 12608 | 8sPWscz3SICG1D8t0001 | xenium/2.0.0/Xenium_V1_humanLung_Cancer_FFPE_o... | None | .zarr | None | spatialdata | 7045222972 | jgalhtHw00CzuZA_jrTygw | 1457 | None | ... | True | False | 2025-12-16 14:14:28.067901+00:00 | 1 | 1 | 1 | 2 | None | None | 40 |
1 rows × 22 columns
Analyze spatial data¶
sdata = all_xenium_data[0].load()
Show code cell output
no parent found for <ome_zarr.reader.Label object at 0x7f4e15a4a540>: None
no parent found for <ome_zarr.reader.Label object at 0x7f4e15abe840>: None
→ transferred: Artifact(uid='8sPWscz3SICG1D8t0001'), Storage(uid='D9BilDV2')
Spatial data datasets stored as SpatialData objects can easily be examined and analyzed through the SpatialData framework, squidpy, and scanpy.
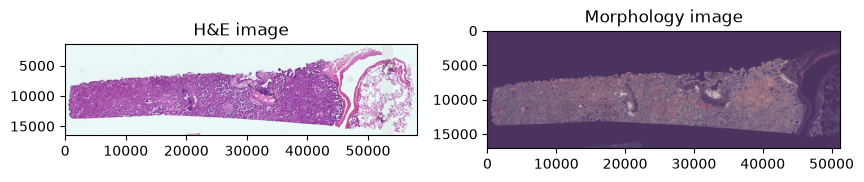
We use spatialdata-plot to get an overview of the dataset:
axes = plt.subplots(1, 2, figsize=(10, 10))[1].flatten()
sdata.pl.render_images("he_image", scale="scale4").pl.show(
ax=axes[0], title="H&E image"
)
sdata.pl.render_images("morphology_focus", scale="scale4").pl.show(
ax=axes[1], title="Morphology image"
)
WARNING render_images: Channel 'dummy' has a constant value (0).
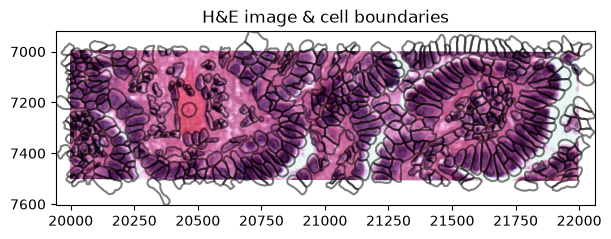
We can visualize the segmentations masks by rendering the shapes from the SpatialData object:
def crop0(x):
return sd.bounding_box_query(
x,
min_coordinate=[20000, 7000],
max_coordinate=[22000, 7500],
axes=("x", "y"),
target_coordinate_system="global",
)
crop0(sdata).pl.render_images("he_image", scale="scale2").pl.render_shapes(
"cell_boundaries", fill_alpha=0, outline_alpha=0.5
).pl.show(
figsize=(6, 3), title="H&E image & cell boundaries", coordinate_systems="global"
)
For any Xenium analysis we would use the AnnData object, which contains the count matrix, cell and gene annotations.
It is stored in the spatialdata.tables slot:
adata = sdata.tables["table"]
adata
Show code cell output
AnnData object with n_obs × n_vars = 154472 × 377
obs: 'cell_id', 'transcript_counts', 'control_probe_counts', 'control_codeword_counts', 'unassigned_codeword_counts', 'deprecated_codeword_counts', 'total_counts', 'cell_area', 'nucleus_area', 'region', 'z_level', 'nucleus_count', 'cell_labels', 'n_genes_by_counts', 'log1p_n_genes_by_counts', 'log1p_total_counts', 'pct_counts_in_top_10_genes', 'pct_counts_in_top_20_genes', 'pct_counts_in_top_50_genes', 'pct_counts_in_top_150_genes', 'n_counts', 'leiden'
var: 'gene_ids', 'feature_types', 'genome', 'n_cells_by_counts', 'mean_counts', 'log1p_mean_counts', 'pct_dropout_by_counts', 'total_counts', 'log1p_total_counts', 'n_cells'
uns: 'umap', 'log1p', 'leiden', 'pca', 'neighbors', 'spatialdata_attrs'
obsm: 'X_pca', 'X_umap', 'spatial'
varm: 'PCs'
obsp: 'distances', 'connectivities'
layers: 'counts', None (.X)
adata.obs
Show code cell output
| cell_id | transcript_counts | control_probe_counts | control_codeword_counts | unassigned_codeword_counts | deprecated_codeword_counts | total_counts | cell_area | nucleus_area | region | ... | cell_labels | n_genes_by_counts | log1p_n_genes_by_counts | log1p_total_counts | pct_counts_in_top_10_genes | pct_counts_in_top_20_genes | pct_counts_in_top_50_genes | pct_counts_in_top_150_genes | n_counts | leiden | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | aaaaficg-1 | 19 | 0 | 0 | 0 | 0 | 19.0 | 49.130002 | 21.268595 | cell_circles | ... | 2 | 15 | 2.772589 | 2.995732 | 73.684211 | 100.000000 | 100.000000 | 100.0 | 19.0 | 2 |
| 2 | aaabbaka-1 | 53 | 0 | 0 | 0 | 0 | 53.0 | 119.618911 | 74.778753 | cell_circles | ... | 3 | 38 | 3.663562 | 3.988984 | 45.283019 | 66.037736 | 100.000000 | 100.0 | 53.0 | 1 |
| 3 | aaabbjoo-1 | 29 | 0 | 0 | 0 | 0 | 29.0 | 94.241097 | 59.109533 | cell_circles | ... | 4 | 18 | 2.944439 | 3.401197 | 72.413793 | 100.000000 | 100.000000 | 100.0 | 29.0 | 0 |
| 4 | aaablchg-1 | 42 | 0 | 0 | 1 | 0 | 42.0 | 120.341411 | 52.426408 | cell_circles | ... | 5 | 33 | 3.526361 | 3.761200 | 45.238095 | 69.047619 | 100.000000 | 100.0 | 42.0 | 1 |
| 5 | aaacaicl-1 | 135 | 0 | 0 | 0 | 0 | 135.0 | 197.965007 | 56.084065 | cell_circles | ... | 6 | 62 | 4.143135 | 4.912655 | 50.370370 | 68.888889 | 91.111111 | 100.0 | 135.0 | 1 |
| ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... |
| 162244 | oipogjpd-1 | 12 | 0 | 0 | 0 | 0 | 12.0 | 6.818594 | 6.818594 | cell_circles | ... | 162245 | 12 | 2.564949 | 2.564949 | 83.333333 | 100.000000 | 100.000000 | 100.0 | 12.0 | 11 |
| 162245 | oipoiane-1 | 11 | 0 | 0 | 0 | 0 | 11.0 | 13.140469 | 13.140469 | cell_circles | ... | 162246 | 9 | 2.302585 | 2.484907 | 100.000000 | 100.000000 | 100.000000 | 100.0 | 11.0 | 2 |
| 162246 | oipolbma-1 | 10 | 0 | 0 | 0 | 0 | 10.0 | 34.634845 | 8.940938 | cell_circles | ... | 162247 | 10 | 2.397895 | 2.397895 | 100.000000 | 100.000000 | 100.000000 | 100.0 | 10.0 | 5 |
| 162247 | oippajff-1 | 13 | 0 | 0 | 0 | 0 | 13.0 | 33.731720 | 5.644531 | cell_circles | ... | 162248 | 13 | 2.639057 | 2.639057 | 76.923077 | 100.000000 | 100.000000 | 100.0 | 13.0 | 2 |
| 162248 | ojaacmoj-1 | 11 | 0 | 0 | 0 | 0 | 11.0 | 14.540313 | 14.540313 | cell_circles | ... | 162249 | 10 | 2.397895 | 2.484907 | 100.000000 | 100.000000 | 100.000000 | 100.0 | 11.0 | 7 |
154472 rows × 22 columns
Quality control metrics can be calculated using scanpy.pp.calculate_qc_metrics.
Cells and genes can be filtered based on minimum thresholds using scanpy.pp.filter_cells and scanpy.pp.filter_genes.
For normalization and dimensionality reduction, standard scanpy workflows include:
scanpy.pp.normalize_totalfor library size normalizationscanpy.pp.log1pfor log transformationscanpy.pp.pcafor principal component analysisscanpy.pp.neighborsfor neighborhood graph computation
For this analysis, we use precomputed Leiden clustering results rather than running the full preprocessing pipeline. For a full tutorial on how to perform analysis of Xenium data, we refer to squidpy’s Xenium tutorial.
Visualize annotation on UMAP and spatial coordinates:
sc.pl.umap(
adata,
color=[
"total_counts",
"n_genes_by_counts",
"leiden",
],
wspace=0.1,
)
Show code cell output

sq.pl.spatial_scatter(
adata,
library_id="spatial",
shape=None,
color=[
"leiden",
],
)

ln.finish()
Show code cell output
→ finished Run('8CIGOdTiMKm9uptO') after 46s at 2026-08-03 16:51:10 UTC