Arc Virtual Cell Atlas 
 ¶
¶
With 2.5B expression profiles that map to about 600M cells, the Arc Virtual Cell Atlas is the world’s largest collection of uniformly processed scRNA-seq datasets. Arc distributes the atlas as 460k parquet and h5ad files totaling 41TB on Google Cloud Storage, see github.com/ArcInstitute/arc-virtual-cell-atlas. Lamin mirrors the atlas in a database: lamin.ai/laminlabs/arc-virtual-cell-atlas.
If you use the data academically, please cite the original publications, Youngblut et al. (2025)[1] and Zhang et al. (2025).[2]
To query the atlas with lamindb, you have to install it with the GCP (Google Cloud Platform) extra. We also recommend configuring the bionty and pertdb modules.
# pip install 'lamindb[gcp]'
!lamin settings modules set bionty,pertdb
Create the central query object for this instance:
import lamindb as ln
import pyarrow.compute as pc
db = ln.DB("laminlabs/arc-virtual-cell-atlas")
Show code cell output
! using anonymous user (to identify, call: lamin login)
Tahoe-100M¶
Retrieve the fourteen .h5ad datasets of the Tahoe-100M project:
tahoe = db.Project.get(name="Tahoe-100M")
artifacts_tahoe = db.Artifact.filter(projects=tahoe, suffix=".h5ad")
artifacts_tahoe.to_dataframe()
Show code cell output
| uid | key | description | suffix | kind | otype | size | hash | n_files | n_observations | ... | is_latest | is_locked | created_at | branch_id | created_on_id | space_id | storage_id | run_id | schema_id | created_by_id | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| id | |||||||||||||||||||||
| 1375 | BDttiuV3Te8VB0dU0000 | tahoe100M/2025-02-25/h5ad/plate9_filt_Vevo_Tah... | None | .h5ad | dataset | AnnData | 18791302576 | 4kHbVbmreg6akW6ZgsjxaA | None | 5866669.0 | ... | True | False | 2025-02-25 23:22:22.759201+00:00 | 1 | 1 | 1 | 2 | 1 | 3 | 1 |
| 1374 | czC19UpUEszVH2bU0000 | tahoe100M/2025-02-25/h5ad/plate8_filt_Vevo_Tah... | None | .h5ad | dataset | AnnData | 30390935958 | ilAzEPIh4FlDeTFaJ1dILw | None | 8880979.0 | ... | True | False | 2025-02-25 23:22:22.387666+00:00 | 1 | 1 | 1 | 2 | 1 | 3 | 1 |
| 1373 | DC5cacdJr1VoEXnl0000 | tahoe100M/2025-02-25/h5ad/plate7_filt_Vevo_Tah... | None | .h5ad | dataset | AnnData | 16514746341 | NOS4MY6eYYPOnAB8ViyWYg | None | 5692117.0 | ... | True | False | 2025-02-25 23:22:22.009157+00:00 | 1 | 1 | 1 | 2 | 1 | 3 | 1 |
| 1372 | aAHQ3zbD7n1asyYr0000 | tahoe100M/2025-02-25/h5ad/plate6_filt_Vevo_Tah... | None | .h5ad | dataset | AnnData | 28934897078 | NYvQEqVClziHm0ozWhOw1w | None | 7545393.0 | ... | True | False | 2025-02-25 23:22:21.629962+00:00 | 1 | 1 | 1 | 2 | 1 | 3 | 1 |
| 1371 | EZATJLC4jE7pmwo40000 | tahoe100M/2025-02-25/h5ad/plate5_filt_Vevo_Tah... | None | .h5ad | dataset | AnnData | 19763140865 | VMBKFzOI5cj7UC1UDENP4A | None | 6419498.0 | ... | True | False | 2025-02-25 23:22:21.255154+00:00 | 1 | 1 | 1 | 2 | 1 | 3 | 1 |
| 1370 | tKTeff0ugWqAm4P70000 | tahoe100M/2025-02-25/h5ad/plate4_filt_Vevo_Tah... | None | .h5ad | dataset | AnnData | 23292672278 | BkBXznbSovNWXtzPFITPcQ | None | 7004356.0 | ... | True | False | 2025-02-25 23:22:20.879928+00:00 | 1 | 1 | 1 | 2 | 1 | 3 | 1 |
| 1369 | XVSrkq9pyF1OBLgG0000 | tahoe100M/2025-02-25/h5ad/plate3_filt_Vevo_Tah... | None | .h5ad | dataset | AnnData | 13173722269 | Jnrt7DaSUCGn8D8LS2itaw | None | 4705402.0 | ... | True | False | 2025-02-25 23:22:20.497965+00:00 | 1 | 1 | 1 | 2 | 1 | 3 | 1 |
| 1368 | ZFeVfd0ugAHeWCxm0000 | tahoe100M/2025-02-25/h5ad/plate2_filt_Vevo_Tah... | None | .h5ad | dataset | AnnData | 29037152127 | usxviuqGbuw0RYnECCVCWw | None | 8064658.0 | ... | True | False | 2025-02-25 23:22:20.113956+00:00 | 1 | 1 | 1 | 2 | 1 | 3 | 1 |
| 1367 | aJIqo7bNyJAs9z0r0000 | tahoe100M/2025-02-25/h5ad/plate1_filt_Vevo_Tah... | None | .h5ad | dataset | AnnData | 19070623904 | 9iCNcouMqfNS3HA/2GUWOA | None | 5481420.0 | ... | True | False | 2025-02-25 23:22:19.737995+00:00 | 1 | 1 | 1 | 2 | 1 | 3 | 1 |
| 1366 | vn5cUJCHbjpPPsZx0000 | tahoe100M/2025-02-25/h5ad/plate14_filt_Vevo_Ta... | None | .h5ad | dataset | AnnData | 22427932564 | FrnStRehP16siRGG35ou+g | None | 6518806.0 | ... | True | False | 2025-02-25 23:22:19.357999+00:00 | 1 | 1 | 1 | 2 | 1 | 3 | 1 |
| 1365 | 9L9HZ55HqUL0aqaR0000 | tahoe100M/2025-02-25/h5ad/plate13_filt_Vevo_Ta... | None | .h5ad | dataset | AnnData | 28071589885 | RKOiaay+CHvv+Ukk/N+28A | None | 8501658.0 | ... | True | False | 2025-02-25 23:22:18.977981+00:00 | 1 | 1 | 1 | 2 | 1 | 3 | 1 |
| 1364 | S2h2rPLCaUhZAM9u0000 | tahoe100M/2025-02-25/h5ad/plate12_filt_Vevo_Ta... | None | .h5ad | dataset | AnnData | 37495736876 | VjAkWVFGVpzAMi9Innusuw | None | 10487057.0 | ... | True | False | 2025-02-25 23:22:18.600910+00:00 | 1 | 1 | 1 | 2 | 1 | 3 | 1 |
| 1363 | omn7JStfJMzy8m6O0000 | tahoe100M/2025-02-25/h5ad/plate11_filt_Vevo_Ta... | None | .h5ad | dataset | AnnData | 23230802756 | N2mzoYlMLEl6PdecaYyDvw | None | 7435869.0 | ... | True | False | 2025-02-25 23:22:18.229629+00:00 | 1 | 1 | 1 | 2 | 1 | 3 | 1 |
| 1362 | 56uA9lPPmJ4zLUcr0000 | tahoe100M/2025-02-25/h5ad/plate10_filt_Vevo_Ta... | None | .h5ad | dataset | AnnData | 26536400717 | j1FXsX7hs7u+eBqnWnmNHw | None | 8044908.0 | ... | True | False | 2025-02-25 23:22:17.849980+00:00 | 1 | 1 | 1 | 2 | 1 | 3 | 1 |
| 15 | 3Yl20zyG926CkvP50000 | tahoe100M/2025-02-25/tutorial/plate3_2k-obs.h5ad | None | .h5ad | dataset | AnnData | 7253540 | vv16qryJsVY98jDBqhkr9w | None | 2000.0 | ... | True | False | 2025-02-25 19:31:01.255128+00:00 | 1 | 1 | 1 | 2 | 1 | 3 | 1 |
15 rows × 22 columns
See the schema and annotations of the first dataset:
artifact1 = artifacts_tahoe[0]
artifact1.describe()
Show code cell output
Artifact: tahoe100M/2025-02-25/h5ad/plate10_filt_Vevo_Tahoe100M_WServicesFrom_ParseGigalab.h5ad (0000) ├── uid: 56uA9lPPmJ4zLUcr0000 run: 0xj4zui (register-tahoe100.ipynb) │ kind: dataset otype: AnnData │ hash: j1FXsX7hs7u+eBqnWnmNHw size: 24.7 GB │ branch: main space: all │ created_at: 2025-02-25 23:22:17 UTC created_by: sunnyosun │ n_observations: 8044908.0 schema: tahoe100_anndata_schema ├── storage/path: │ gs://arc-institute-virtual-cell-atlas/tahoe100M/2025-02-25/h5ad/plate10_filt_Vevo_Tahoe100M_WServicesFrom_Parse │ Gigalab.h5ad ├── Dataset features │ ├── var (62710.0 bionty.Gene.sta… │ │ ANKIB1 float │ │ C1orf112 float │ │ CFH float │ │ CFTR float │ │ CYP51A1 float │ │ DPM1 float │ │ ENPP4 float │ │ FGR float │ │ FUCA2 float │ │ GCLC float │ │ KRIT1 float │ │ LAS1L float │ │ NFYA float │ │ NIPAL3 float │ │ RAD52 float │ │ SCYL3 float │ │ SEMA3F float │ │ STPG1 float │ │ TNMD float │ │ TSPAN6 float │ └── obs (16.0) │ BARCODE str │ G2M_score float │ S_score float │ cell_line bionty.CellLine A-172, A-427, A498, A549, AN3 CA, AsPC… │ cell_name bionty.CellLine A-172, A-427, A498, A549, AN3 CA, AsPC… │ drug pertdb.Compound 5-Azacytidine, 5-Fluorouracil, Abirate… │ drugname_drugconc pertdb.CompoundPerturbation [('5-Azacytidine', 0.05, 'uM')], [('5-… │ gene_count int │ mread_count int │ pass_filter ULabel[yMABN5Dr] full, minimal │ pcnt_mito float │ phase ULabel[kTzOKZ54] G1, G2M, S │ plate ULabel[SjVCuE2Q] plate10 │ sample str │ sublibrary str │ tscp_count int └── Labels └── .ulabels ULabel plate10, G1, G2M, S, full, minimal .projects Project Tahoe-100M .organisms bionty.Organism human .cell_lines bionty.CellLine NCI-H1573, NCI-H460, hTERT-HPNE, SW48,… .compounds pertdb.Compound Bestatin (hydrochloride), Ataluren, Ca… .compound_perturbations pertdb.CompoundPerturbation [('Bestatin (hydrochloride)', 0.05, 'u…
You can download an .h5ad into your local cache, load it into memory, or open it for streaming:
local_filepath = artifact1.cache() # sync into cache
adata = artifact1.load() # sync into cache and load into memory
with artifact1.open() as adata: # open for streaming
...
You can query the CellLine ontology, the Compound, and the CompoundPerturbation registries via their relationship to Artifact. You’ll find 50 cell lines:
db.bionty.CellLine.filter(artifacts__in=artifacts_tahoe).distinct().to_dataframe()
Show code cell output
! truncated query result to limit=20 CellLine objects
| uid | name | ontology_id | abbr | synonyms | description | is_locked | created_at | branch_id | created_on_id | space_id | created_by_id | run_id | source_id | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| id | ||||||||||||||
| 50 | 7VaGVBNBdVEmdf | NCI-H596 | CVCL_1571 | None | H596|H-596|NCI-HUT-596|NCIH596 | Part of: Cancer Dependency Map project (DepMap... | False | 2025-02-25 22:20:20.217993+00:00 | 1 | 1 | 1 | 1 | 3 | 120 |
| 49 | 7dL2LJjx9iARSo | NCI-H661 | CVCL_1577 | None | H661|H-661|NCIH661 | Part of: AKT genetic alteration cell panel (AT... | False | 2025-02-25 22:20:20.217993+00:00 | 1 | 1 | 1 | 1 | 3 | 120 |
| 48 | J5Ylm8TVnGfDCh | NCI-H2122 | CVCL_1531 | None | H2122|H-2122|NCIH2122 | Part of: Cancer Dependency Map project (DepMap... | False | 2025-02-25 22:20:20.217993+00:00 | 1 | 1 | 1 | 1 | 3 | 120 |
| 47 | 6O2MPQMm2fYmHx | A-427 | CVCL_1055 | None | A427|A427N | Part of: Cancer Dependency Map project (DepMap... | False | 2025-02-25 22:20:20.217993+00:00 | 1 | 1 | 1 | 1 | 3 | 120 |
| 46 | 219BOZMeuZbXcN | SW 1088 | CVCL_1715 | None | SW-1088|SW 1088 | Part of: Cancer Dependency Map project (DepMap... | False | 2025-02-25 22:20:20.217993+00:00 | 1 | 1 | 1 | 1 | 3 | 120 |
| 45 | 2eQosYlsjKld2O | CHP-212 | CVCL_1125 | None | CHP 212|CHP212|NB9|NB-9|Children's Hospital of... | Part of: Cancer Dependency Map project (DepMap... | False | 2025-02-25 22:20:20.217993+00:00 | 1 | 1 | 1 | 1 | 3 | 120 |
| 44 | 5JZNtoDJwHkHJl | AsPC-1 | CVCL_0152 | None | AsPc-1|Aspc-1|ASPC-1|As-PC1|ASPC1|AsPC1|Aspc1|... | Part of: Cancer Dependency Map project (DepMap... | False | 2025-02-25 22:20:20.217993+00:00 | 1 | 1 | 1 | 1 | 3 | 120 |
| 43 | 3Oz9gRsuI8mZjq | HepG2/C3A | CVCL_1098 | None | HepG2/C3A|Hep G2/C3A|C3A | Group: Patented cell line. Part of: Cancer Dep... | False | 2025-02-25 22:20:20.217993+00:00 | 1 | 1 | 1 | 1 | 3 | 120 |
| 42 | 1K1CzNSiUR7v5M | C32 | CVCL_1097 | None | C-32|C32-mel|C32 mel|C32r | Part of: Cancer Dependency Map project (DepMap... | False | 2025-02-25 22:20:20.217993+00:00 | 1 | 1 | 1 | 1 | 3 | 120 |
| 41 | 4QH2SpWAsqvVcp | NCI-H2030 | CVCL_1517 | None | H2030|H-2030|NCIH2030 | Part of: Cancer Dependency Map project (DepMap... | False | 2025-02-25 22:20:20.217993+00:00 | 1 | 1 | 1 | 1 | 3 | 120 |
| 40 | vEfTp1Hk8gZOig | A549 | CVCL_0023 | None | A 549|A549|NCI-A549|A549/ATCC|A549 ATCC|A549AT... | Part of: Cancer Dependency Map project (DepMap... | False | 2025-02-25 22:20:20.217993+00:00 | 1 | 1 | 1 | 1 | 3 | 120 |
| 39 | 39rNVaPPNTJKGZ | SK-MEL-2 | CVCL_0069 | None | SK-Mel-2|SK-Mel 2|SK-mel-2|SK-MEL2|SK.MEL.2|SK... | Part of: Cancer Dependency Map project (DepMap... | False | 2025-02-25 22:20:20.217993+00:00 | 1 | 1 | 1 | 1 | 3 | 120 |
| 38 | 1lhqeW2vEMefxI | RPMI-7951 | CVCL_1666 | None | RPMI 7951|RPMI7951|Roswell Park Memorial Insti... | Part of: BRAF genetic alteration cell panel (A... | False | 2025-02-25 22:20:20.217993+00:00 | 1 | 1 | 1 | 1 | 3 | 120 |
| 37 | 5lqReFKRVIe0Mh | AN3 CA | CVCL_0028 | None | AN3_CA|AN3 CA|AN3 Ca|AN3CA|AN-3|AN3|Acanthosis... | Part of: Cancer Dependency Map project (DepMap... | False | 2025-02-25 22:20:20.217993+00:00 | 1 | 1 | 1 | 1 | 3 | 120 |
| 36 | 4hyU9oFufh0bK1 | BT-474 | CVCL_0179 | None | Bt-474|BT474 | Part of: AKT genetic alteration cell panel (AT... | False | 2025-02-25 22:20:20.217993+00:00 | 1 | 1 | 1 | 1 | 3 | 120 |
| 35 | 5XupQdHONULNf4 | COLO 205 | CVCL_0218 | None | Colo 205|CoLo 205|COLO-205|Colo-205|COLO.205|C... | Part of: AstraZeneca Colorectal cell line (AZC... | False | 2025-02-25 22:20:20.217993+00:00 | 1 | 1 | 1 | 1 | 3 | 120 |
| 34 | 63ZWvcHVlrgadr | HCT15 | CVCL_0292 | None | HCT-15|HCT.15|HCT15 | Part of: Cancer Dependency Map project (DepMap... | False | 2025-02-25 22:20:20.217993+00:00 | 1 | 1 | 1 | 1 | 3 | 120 |
| 33 | 1mLuzzowHZrJ1x | HEC-1-A | CVCL_0293 | None | Hec-1-A|HEC-1A|HEC1-A|HEC1A|Hec1A | Part of: Cancer Dependency Map project (DepMap... | False | 2025-02-25 22:20:20.217993+00:00 | 1 | 1 | 1 | 1 | 3 | 120 |
| 32 | 1CdEQ5dJ3wSnz0 | LS 180 | CVCL_0397 | None | LS-180|LS 180|Laboratory of Surgery 180 | Group: Patented cell line. Part of: AstraZenec... | False | 2025-02-25 22:20:20.217993+00:00 | 1 | 1 | 1 | 1 | 3 | 120 |
| 31 | 5ewuYry0FV3ZfQ | Panc 03.27 | CVCL_1635 | None | Panc 3.27|Panc-03.27|PANC-03-27|Panc_03_27|Pan... | Part of: Cancer Dependency Map project (DepMap... | False | 2025-02-25 22:20:20.217993+00:00 | 1 | 1 | 1 | 1 | 3 | 120 |
380 compounds:
db.pertdb.Compound.filter(artifacts__in=artifacts_tahoe).distinct().to_dataframe()
Show code cell output
! truncated query result to limit=20 Compound objects
| uid | ontology_id | abbr | synonyms | description | name | type | chembl_id | smiles | canonical_smiles | ... | molformula | moa | is_locked | created_at | branch_id | created_on_id | space_id | created_by_id | run_id | source_id | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| id | |||||||||||||||||||||
| 380 | JRDV3CsZkr3Yuu | None | None | None | Cyclooxygenase inhibitor | Tolmetin | None | None | None | None | ... | None | None | False | 2025-02-25 22:48:58.568677+00:00 | 1 | 1 | 1 | 1 | 3 | None |
| 379 | x3BfjX6JwJhYJu | None | None | None | Retinoic receptor agonist | Peretinoin | None | None | None | None | ... | None | None | False | 2025-02-25 22:48:58.568677+00:00 | 1 | 1 | 1 | 1 | 3 | None |
| 378 | 18PJ8Lu8ZS7C48 | None | None | None | None | Niclosamide (olamine) | None | None | None | None | ... | None | None | False | 2025-02-25 22:48:58.568677+00:00 | 1 | 1 | 1 | 1 | 3 | None |
| 377 | 5yeFtKHyuB3qyk | None | None | None | Androgen receptor antagonist | Apalutamide | None | None | None | None | ... | None | None | False | 2025-02-25 22:48:58.568677+00:00 | 1 | 1 | 1 | 1 | 3 | None |
| 376 | 2AEfoFfqFY4Tom | None | None | None | None | Mifepristone | None | None | None | None | ... | None | None | False | 2025-02-25 22:48:58.568677+00:00 | 1 | 1 | 1 | 1 | 3 | None |
| 375 | 7TxN4mX3JsIySH | None | None | None | None | Eplerenone | None | None | None | None | ... | None | None | False | 2025-02-25 22:48:58.568677+00:00 | 1 | 1 | 1 | 1 | 3 | None |
| 374 | 65yWmo3FuI81Bp | None | None | None | None | Macitentan | None | None | None | None | ... | None | None | False | 2025-02-25 22:48:58.568677+00:00 | 1 | 1 | 1 | 1 | 3 | None |
| 373 | 4npiPmNGZSDQLH | None | None | None | None | Tranilast | None | None | None | None | ... | None | None | False | 2025-02-25 22:48:58.568677+00:00 | 1 | 1 | 1 | 1 | 3 | None |
| 372 | 46hvCmCeQXlHS3 | None | None | None | Microtubule inhibitor | Docetaxel (Trihydrate) | None | None | None | None | ... | None | None | False | 2025-02-25 22:48:58.568677+00:00 | 1 | 1 | 1 | 1 | 3 | None |
| 371 | 6e9zB661U1eEIb | None | None | None | None | Arbutin | None | None | None | None | ... | None | None | False | 2025-02-25 22:48:58.568677+00:00 | 1 | 1 | 1 | 1 | 3 | None |
| 370 | 4DOayww0V9b2Vs | None | None | None | Multi-TK inhibitor | Sulfatinib | None | None | None | None | ... | None | None | False | 2025-02-25 22:48:58.568677+00:00 | 1 | 1 | 1 | 1 | 3 | None |
| 369 | aAg6I5NZssKbUD | None | None | None | None | Mozavaptan | None | None | None | None | ... | None | None | False | 2025-02-25 22:48:58.568677+00:00 | 1 | 1 | 1 | 1 | 3 | None |
| 368 | 6NymH8sy4mIM43 | None | None | None | DNA synthesis/repair inhibitor | Clofarabine | None | None | None | None | ... | None | None | False | 2025-02-25 22:48:58.568677+00:00 | 1 | 1 | 1 | 1 | 3 | None |
| 367 | 6NxdkGLlz722L4 | None | None | None | None | Imiquimod (maleate) | None | None | None | None | ... | None | None | False | 2025-02-25 22:48:58.568677+00:00 | 1 | 1 | 1 | 1 | 3 | None |
| 366 | LNRydFsxWv1OiZ | None | None | None | Glucose transporter inhibitor | Dapagliflozin ((2S)-1,2-propanediol, hydrate) | None | None | None | None | ... | None | None | False | 2025-02-25 22:48:58.568677+00:00 | 1 | 1 | 1 | 1 | 3 | None |
| 365 | 1YnTm1CfbADqQp | None | None | None | None | Glycyrrhizic acid | None | None | None | None | ... | None | None | False | 2025-02-25 22:48:58.568677+00:00 | 1 | 1 | 1 | 1 | 3 | None |
| 364 | 1surTvrPkwS2mF | None | None | None | None | Menadione | None | None | None | None | ... | None | None | False | 2025-02-25 22:48:58.568677+00:00 | 1 | 1 | 1 | 1 | 3 | None |
| 363 | 6rFH03Kak2P7t1 | None | None | None | DNA synthesis/repair inhibitor | Doxorubicin (hydrochloride) | None | None | None | None | ... | None | None | False | 2025-02-25 22:48:58.568677+00:00 | 1 | 1 | 1 | 1 | 3 | None |
| 362 | 423Hz3ZAC2KnIf | None | None | None | Adrenoceptor agonist | Dexmedetomidine | None | None | None | None | ... | None | None | False | 2025-02-25 22:48:58.568677+00:00 | 1 | 1 | 1 | 1 | 3 | None |
| 361 | 5C8fjLZFHyp8JP | None | None | None | None | Rifaximin | None | None | None | None | ... | None | None | False | 2025-02-25 22:48:58.568677+00:00 | 1 | 1 | 1 | 1 | 3 | None |
20 rows × 22 columns
1,138 perturbations:
db.pertdb.CompoundPerturbation.filter(artifacts__in=artifacts_tahoe).distinct().to_dataframe()
Show code cell output
! truncated query result to limit=20 CompoundPerturbation objects
| uid | abbr | synonyms | description | name | concentration | concentration_unit | duration | is_locked | created_at | branch_id | created_on_id | space_id | created_by_id | run_id | source_id | compound_id | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| id | |||||||||||||||||
| 1138 | 37rn9xx8Vzca2L | None | None | None | [('Tolmetin', 5.0, 'uM')] | 5.0 | uM | None | False | 2025-02-25 22:59:15.764901+00:00 | 1 | 1 | 1 | 1 | 3 | None | 380 |
| 1137 | 5FRNvnyvkZokCn | None | None | None | [('Peretinoin', 5.0, 'uM')] | 5.0 | uM | None | False | 2025-02-25 22:59:15.764901+00:00 | 1 | 1 | 1 | 1 | 3 | None | 379 |
| 1136 | 6Wdgxi67CDiFF1 | None | None | None | [('Niclosamide (olamine)', 5.0, 'uM')] | 5.0 | uM | None | False | 2025-02-25 22:59:15.764901+00:00 | 1 | 1 | 1 | 1 | 3 | None | 378 |
| 1135 | ZYlNIuNh7HVszg | None | None | None | [('Apalutamide', 5.0, 'uM')] | 5.0 | uM | None | False | 2025-02-25 22:59:15.764901+00:00 | 1 | 1 | 1 | 1 | 3 | None | 377 |
| 1134 | 17Tb1OMJxUcytA | None | None | None | [('Mifepristone', 5.0, 'uM')] | 5.0 | uM | None | False | 2025-02-25 22:59:15.764901+00:00 | 1 | 1 | 1 | 1 | 3 | None | 376 |
| 1133 | 4sSrMIoDWXJOSu | None | None | None | [('Eplerenone', 5.0, 'uM')] | 5.0 | uM | None | False | 2025-02-25 22:59:15.764901+00:00 | 1 | 1 | 1 | 1 | 3 | None | 375 |
| 1132 | l4OnyeRYK2ImQf | None | None | None | [('Macitentan', 5.0, 'uM')] | 5.0 | uM | None | False | 2025-02-25 22:59:15.764901+00:00 | 1 | 1 | 1 | 1 | 3 | None | 374 |
| 1131 | 6PDvhrPAUfBluF | None | None | None | [('Tranilast', 5.0, 'uM')] | 5.0 | uM | None | False | 2025-02-25 22:59:15.764901+00:00 | 1 | 1 | 1 | 1 | 3 | None | 373 |
| 1130 | 7laTXVbGocbsGH | None | None | None | [('Docetaxel (Trihydrate)', 5.0, 'uM')] | 5.0 | uM | None | False | 2025-02-25 22:59:15.764901+00:00 | 1 | 1 | 1 | 1 | 3 | None | 372 |
| 1129 | gxlnwuLJUpf3iB | None | None | None | [('Arbutin', 5.0, 'uM')] | 5.0 | uM | None | False | 2025-02-25 22:59:15.764901+00:00 | 1 | 1 | 1 | 1 | 3 | None | 371 |
| 1128 | 7ah0b02sGULYuN | None | None | None | [('Sulfatinib', 5.0, 'uM')] | 5.0 | uM | None | False | 2025-02-25 22:59:15.764901+00:00 | 1 | 1 | 1 | 1 | 3 | None | 370 |
| 1127 | Tl6edhsU5mmmWY | None | None | None | [('Adagrasib', 5.0, 'uM')] | 5.0 | uM | None | False | 2025-02-25 22:59:15.764901+00:00 | 1 | 1 | 1 | 1 | 3 | None | 94 |
| 1126 | 7RfxE7Vn84jTT6 | None | None | None | [('Mozavaptan', 5.0, 'uM')] | 5.0 | uM | None | False | 2025-02-25 22:59:15.764901+00:00 | 1 | 1 | 1 | 1 | 3 | None | 369 |
| 1125 | 6FojRhBrzl4puB | None | None | None | [('Clofarabine', 5.0, 'uM')] | 5.0 | uM | None | False | 2025-02-25 22:59:15.764901+00:00 | 1 | 1 | 1 | 1 | 3 | None | 368 |
| 1124 | 5TQfCYyXidZl08 | None | None | None | [('Imiquimod (maleate)', 5.0, 'uM')] | 5.0 | uM | None | False | 2025-02-25 22:59:15.764901+00:00 | 1 | 1 | 1 | 1 | 3 | None | 367 |
| 1123 | 5DfQLJdjmtXcW1 | None | None | None | [('Dapagliflozin ((2S)-1,2-propanediol, hydrat... | 5.0 | uM | None | False | 2025-02-25 22:59:15.764901+00:00 | 1 | 1 | 1 | 1 | 3 | None | 366 |
| 1122 | 1jIX1b41fJwHcb | None | None | None | [('Glycyrrhizic acid', 5.0, 'uM')] | 5.0 | uM | None | False | 2025-02-25 22:59:15.764901+00:00 | 1 | 1 | 1 | 1 | 3 | None | 365 |
| 1121 | 5NNuPZQU49frOA | None | None | None | [('Menadione', 5.0, 'uM')] | 5.0 | uM | None | False | 2025-02-25 22:59:15.764901+00:00 | 1 | 1 | 1 | 1 | 3 | None | 364 |
| 1120 | 7iGGYEWsFv2mOs | None | None | None | [('Doxorubicin (hydrochloride)', 5.0, 'uM')] | 5.0 | uM | None | False | 2025-02-25 22:59:15.764901+00:00 | 1 | 1 | 1 | 1 | 3 | None | 363 |
| 1119 | 40csupxNUCRajW | None | None | None | [('Dexmedetomidine', 5.0, 'uM')] | 5.0 | uM | None | False | 2025-02-25 22:59:15.764901+00:00 | 1 | 1 | 1 | 1 | 3 | None | 362 |
Query artifacts based on metadata¶
Let’s find which datasets contain A549 cells perturbed with Piroxicam.
a549 = db.bionty.CellLine.get(name="A549")
piro = db.pertdb.Compound.get(name="Piroxicam")
artifacts_a549_piro = artifacts_tahoe.filter(compounds=piro, cell_lines=a549)
artifacts_a549_piro.to_dataframe()
Show code cell output
| uid | key | description | suffix | kind | otype | size | hash | n_files | n_observations | ... | is_latest | is_locked | created_at | branch_id | created_on_id | space_id | storage_id | run_id | schema_id | created_by_id | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| id | |||||||||||||||||||||
| 1364 | S2h2rPLCaUhZAM9u0000 | tahoe100M/2025-02-25/h5ad/plate12_filt_Vevo_Ta... | None | .h5ad | dataset | AnnData | 37495736876 | VjAkWVFGVpzAMi9Innusuw | None | 10487057.0 | ... | True | False | 2025-02-25 23:22:18.600910+00:00 | 1 | 1 | 1 | 2 | 1 | 3 | 1 |
| 1363 | omn7JStfJMzy8m6O0000 | tahoe100M/2025-02-25/h5ad/plate11_filt_Vevo_Ta... | None | .h5ad | dataset | AnnData | 23230802756 | N2mzoYlMLEl6PdecaYyDvw | None | 7435869.0 | ... | True | False | 2025-02-25 23:22:18.229629+00:00 | 1 | 1 | 1 | 2 | 1 | 3 | 1 |
| 1362 | 56uA9lPPmJ4zLUcr0000 | tahoe100M/2025-02-25/h5ad/plate10_filt_Vevo_Ta... | None | .h5ad | dataset | AnnData | 26536400717 | j1FXsX7hs7u+eBqnWnmNHw | None | 8044908.0 | ... | True | False | 2025-02-25 23:22:17.849980+00:00 | 1 | 1 | 1 | 2 | 1 | 3 | 1 |
3 rows × 22 columns
Stream the dataset content¶
While the artifact metadata tells us which files contain A549 cells and Piroxicam, we use a parquet file to find the exact cells within those files. To this end, we open the metadata file with pyarrow.Dataset:
obs_af = db.Artifact.get(key__endswith="obs_metadata.parquet", projects=tahoe)
obs_af.describe()
Show code cell output
Artifact: tahoe100M/2025-02-25/metadata/obs_metadata.parquet (0000) ├── uid: y1TTR9wbrmZEwpOa0000 run: 0xj4zui (register-tahoe100.ipynb) │ kind: dataset otype: DataFrame │ hash: qEWOpGw9CmQVzaElyMWT1Q size: 2.1 GB │ branch: main space: all │ created_at: 2025-02-25 19:33:42 UTC created_by: sunnyosun │ n_observations: 100648790.0 ├── storage/path: gs://arc-institute-virtual-cell-atlas/tahoe100M/2025-02-25/metadata/obs_metadata.parquet └── Labels └── .ulabels ULabel metadata .projects Project Tahoe-100M .organisms bionty.Organism human
The schema of the parquet file maps to the pyarrow schema:
obs_ds = obs_af.open() # consider using with obs_af.open() as obs_ds
obs_ds.schema
Show code cell output
plate: string
BARCODE_SUB_LIB_ID: string
sample: string
gene_count: int64
tscp_count: int64
mread_count: int64
drugname_drugconc: string
drug: string
cell_line: dictionary<values=string, indices=int8, ordered=0>
sublibrary: string
BARCODE: string
pcnt_mito: float
S_score: double
G2M_score: double
phase: dictionary<values=string, indices=int8, ordered=0>
pass_filter: dictionary<values=string, indices=int8, ordered=0>
cell_name: dictionary<values=string, indices=int8, ordered=0>
__index_level_0__: int64
-- schema metadata --
pandas: '{"index_columns": ["__index_level_0__"], "column_indexes": [{"na' + 2487
Streaming speed
Streaming large parquet and h5ad files from cloud storage crucially depends on where you run your code. It’ll be much faster if you run it in the data center that hosts the data. It’ll typically be prohibitively slow if you run it locally. The gs://arc-institute-virtual-cell-atlas storage location is accessible from any Google Cloud data center in the US with low latency and no egress fees.
If you want to run logic locally, consider caching datasets prior to opening them for streaming via .open():
local_filepath = obs_af.cache() # subsequent obs_af.open() will automatically read from the cache
Let us now query the columns of interest:
filter_expr = (pc.field("cell_name") == a549.name) & (pc.field("drug") == piro.name)
Retrieve the corresponding cells:
plate_cells = obs_df.groupby("plate")["BARCODE_SUB_LIB_ID"].apply(list)
And their counts:
adatas = []
for artifact in artifacts_a549_piro:
plate_name = artifact.features["plate"].name
idxs = plate_cells.get(plate_name)
print(f"loading {len(idxs)} cells from plate {plate_name}")
with artifact.open() as astore:
adata = astore[idxs].to_memory() # can also subset genes here
adatas.append(adata)
# this will print something like this
#> loading 2812 cells from plate plate10
#> ...
# continue with concatenating or other processing of the AnnData objects
Train ML models¶
By applying fast data loaders such as annbatch[3] or scdataset[4] to locally cached arrays, one can achieve loading times of 50k - 80k vectors/second. This is much faster than cloud-based streaming of the array content.
Here we zero-copy transferred the Tahoe-100M datasets into a database for benchmarking different ML data loaders:
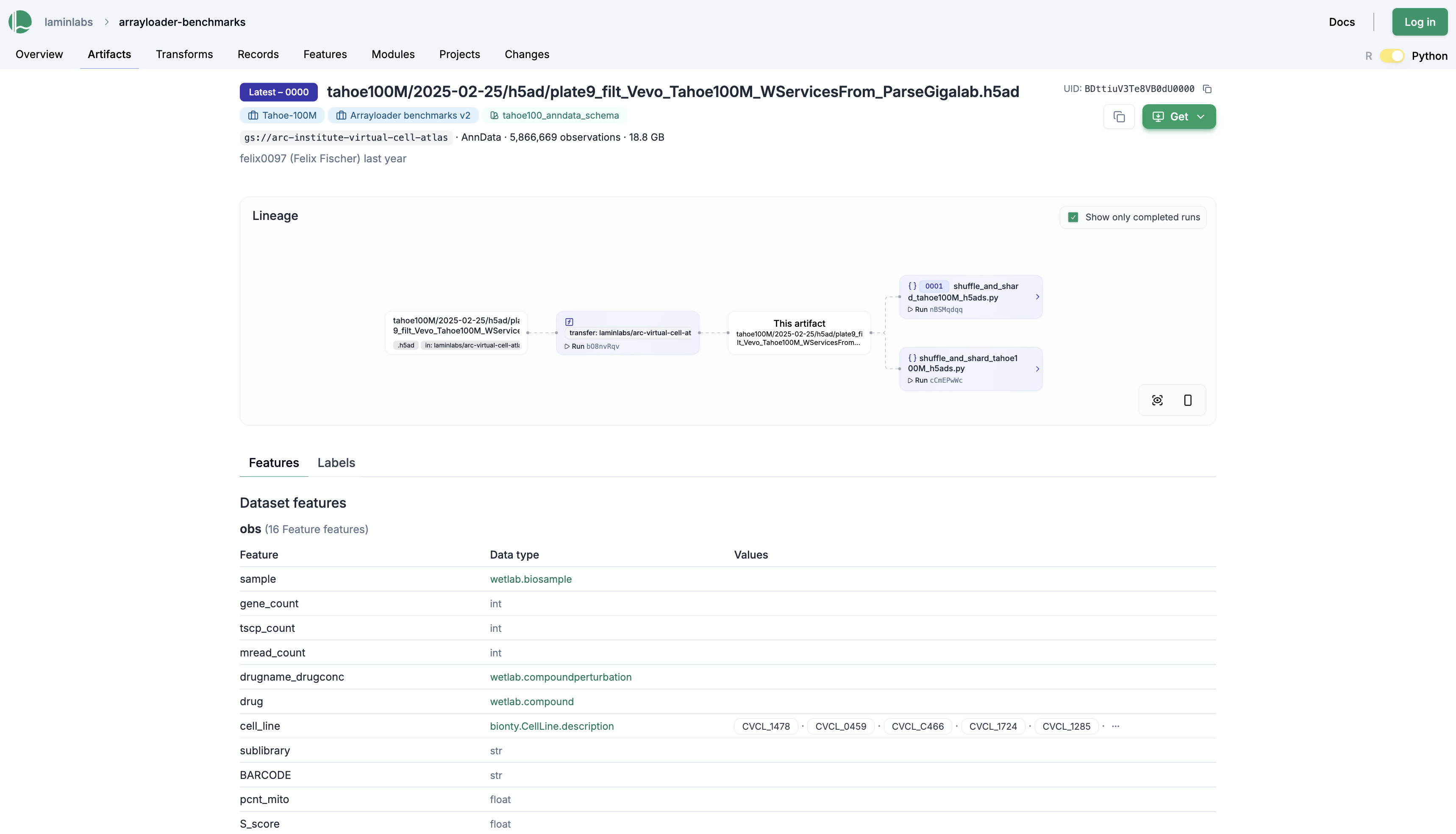
Here is an example for a data loading run that loads these Tahoe-100M datasets from a pre-shuffled .zarr store, obtained as a transformation of the original 14 .h5ad files.
scBaseCount¶
scbase = db.Project.get(name="scBaseCount")
scbase
Show code cell output
Project(uid='vdK00t9DGwHP', is_type=False, name='scBaseCount', description=None, abbr=None, url='https://arcinstitute.org/tools/virtualcellatlas', start_date=None, end_date=None, branch_id=1, created_on_id=1, space_id=1, created_by_id=1, run_id=None, type_id=None, created_at=2025-02-26 16:04:08 UTC, is_locked=False)
Query artifacts based on metadata¶
An exemplary query:
organisms = db.bionty.Organism.lookup()
tissues = db.bionty.Tissue.lookup()
efos = db.bionty.ExperimentalFactor.lookup()
feature_counts = db.ULabel.filter(type__name="STARsolo count features").lookup()
h5ads_brain = db.Artifact.filter(
version_tag="2026-01-12",
suffix=".h5ad",
projects=scbase,
organisms=organisms.human,
ulabels=feature_counts.genefull_ex50pas,
tissues=tissues.brain,
experimental_factors=efos.single_cell,
).order_by("size").distinct()
h5ads_brain.to_dataframe()
Show code cell output
! truncated query result to limit=20 Artifact objects
| uid | key | description | suffix | kind | otype | size | hash | n_files | n_observations | ... | is_latest | is_locked | created_at | branch_id | created_on_id | space_id | storage_id | run_id | schema_id | created_by_id | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| id | |||||||||||||||||||||
| 554817 | qvSEhDQmKxucI3760000 | scbasecount/2026-01-12/h5ad/GeneFull_Ex50pAS/H... | None | .h5ad | dataset | AnnData | 2738016 | 6B9vkGpnXxI9Q23rTDyC9w | None | None | ... | True | False | 2026-05-20 20:13:55.494800+00:00 | 1 | 1 | 1 | 2 | 33 | 62 | 1 |
| 541733 | eYQD5k3PzRRMstxW0001 | scbasecount/2026-01-12/h5ad/GeneFull_Ex50pAS/H... | None | .h5ad | dataset | AnnData | 2878065 | eUQqSyIIYUHwABkh80+tPA | None | None | ... | True | False | 2026-05-20 20:13:55.494800+00:00 | 1 | 1 | 1 | 2 | 33 | 62 | 1 |
| 541705 | SqUgBbn2N6boglmW0001 | scbasecount/2026-01-12/h5ad/GeneFull_Ex50pAS/H... | None | .h5ad | dataset | AnnData | 2972876 | fGuBQlQ0LMWrnjGr4ZaD0Q | None | None | ... | True | False | 2026-05-20 20:13:55.494800+00:00 | 1 | 1 | 1 | 2 | 33 | 62 | 1 |
| 541726 | KVWrMQWjAozuBHz20001 | scbasecount/2026-01-12/h5ad/GeneFull_Ex50pAS/H... | None | .h5ad | dataset | AnnData | 2984182 | UFWF5MNgROBmEWe5PnxbGQ | None | None | ... | True | False | 2026-05-20 20:13:55.494800+00:00 | 1 | 1 | 1 | 2 | 33 | 62 | 1 |
| 541706 | RdS7w8hsDNyD8iKz0001 | scbasecount/2026-01-12/h5ad/GeneFull_Ex50pAS/H... | None | .h5ad | dataset | AnnData | 2998051 | 3ng4kuS5t/GepXCPYI2m6Q | None | None | ... | True | False | 2026-05-20 20:13:55.494800+00:00 | 1 | 1 | 1 | 2 | 33 | 62 | 1 |
| 541707 | zHGkLgEsKwD5dM600001 | scbasecount/2026-01-12/h5ad/GeneFull_Ex50pAS/H... | None | .h5ad | dataset | AnnData | 3020129 | DFRLqyzjNQOw/PPp66G22w | None | None | ... | True | False | 2026-05-20 20:13:55.494800+00:00 | 1 | 1 | 1 | 2 | 33 | 62 | 1 |
| 541724 | aU2UtgUaG4IXGZ3q0001 | scbasecount/2026-01-12/h5ad/GeneFull_Ex50pAS/H... | None | .h5ad | dataset | AnnData | 3061663 | ag/agNEd156c2ncyP4HPCw | None | None | ... | True | False | 2026-05-20 20:13:55.494800+00:00 | 1 | 1 | 1 | 2 | 33 | 62 | 1 |
| 541730 | olRobawASPX65d8R0001 | scbasecount/2026-01-12/h5ad/GeneFull_Ex50pAS/H... | None | .h5ad | dataset | AnnData | 3088141 | IgQ+SeavjSwdRZPHQ9uujQ | None | None | ... | True | False | 2026-05-20 20:13:55.494800+00:00 | 1 | 1 | 1 | 2 | 33 | 62 | 1 |
| 541723 | bRKujvO172sS94BW0001 | scbasecount/2026-01-12/h5ad/GeneFull_Ex50pAS/H... | None | .h5ad | dataset | AnnData | 3102283 | MNJX4U40istgMbt/XV0Xkw | None | None | ... | True | False | 2026-05-20 20:13:55.494800+00:00 | 1 | 1 | 1 | 2 | 33 | 62 | 1 |
| 541725 | wTscUTiAn41DFyz40001 | scbasecount/2026-01-12/h5ad/GeneFull_Ex50pAS/H... | None | .h5ad | dataset | AnnData | 3104274 | +IBcVdXjUhg0u2ILhOCQMg | None | None | ... | True | False | 2026-05-20 20:13:55.494800+00:00 | 1 | 1 | 1 | 2 | 33 | 62 | 1 |
| 541722 | MZE8kJoOvWoB1m050001 | scbasecount/2026-01-12/h5ad/GeneFull_Ex50pAS/H... | None | .h5ad | dataset | AnnData | 3148034 | 5pxMlWY5em71IPUPLGPP5w | None | None | ... | True | False | 2026-05-20 20:13:55.494800+00:00 | 1 | 1 | 1 | 2 | 33 | 62 | 1 |
| 541732 | rZZG71atPzfOKYLr0001 | scbasecount/2026-01-12/h5ad/GeneFull_Ex50pAS/H... | None | .h5ad | dataset | AnnData | 3243289 | fOfaXgAqo0VTrAGjbEKvag | None | None | ... | True | False | 2026-05-20 20:13:55.494800+00:00 | 1 | 1 | 1 | 2 | 33 | 62 | 1 |
| 541721 | SUxUpHMVSy1mvFYI0001 | scbasecount/2026-01-12/h5ad/GeneFull_Ex50pAS/H... | None | .h5ad | dataset | AnnData | 3246360 | dNY/1DTYsLhuhZiWTea27A | None | None | ... | True | False | 2026-05-20 20:13:55.494800+00:00 | 1 | 1 | 1 | 2 | 33 | 62 | 1 |
| 541702 | 7FYIJKkc6RXpQXXC0001 | scbasecount/2026-01-12/h5ad/GeneFull_Ex50pAS/H... | None | .h5ad | dataset | AnnData | 3294428 | zX+l+LxlI6KC/O7uc1Rbcw | None | None | ... | True | False | 2026-05-20 20:13:55.494800+00:00 | 1 | 1 | 1 | 2 | 33 | 62 | 1 |
| 541709 | KXAACADoPpolZA3l0001 | scbasecount/2026-01-12/h5ad/GeneFull_Ex50pAS/H... | None | .h5ad | dataset | AnnData | 3307676 | OA1LDbwg5K+vT/scEOvMzA | None | None | ... | True | False | 2026-05-20 20:13:55.494800+00:00 | 1 | 1 | 1 | 2 | 33 | 62 | 1 |
| 542977 | zNynawwbm5fhAQtL0001 | scbasecount/2026-01-12/h5ad/GeneFull_Ex50pAS/H... | None | .h5ad | dataset | AnnData | 3459556 | /AfVn1ecRgZUMSO2APtMVw | None | None | ... | True | False | 2026-05-20 20:13:55.494800+00:00 | 1 | 1 | 1 | 2 | 33 | 62 | 1 |
| 541731 | 20H5WHxQBQDOQSyf0001 | scbasecount/2026-01-12/h5ad/GeneFull_Ex50pAS/H... | None | .h5ad | dataset | AnnData | 3474227 | niAL47ujFCANpIeJt8/XBQ | None | None | ... | True | False | 2026-05-20 20:13:55.494800+00:00 | 1 | 1 | 1 | 2 | 33 | 62 | 1 |
| 543554 | 7bCCJl948w61tmoK0001 | scbasecount/2026-01-12/h5ad/GeneFull_Ex50pAS/H... | None | .h5ad | dataset | AnnData | 3720165 | kRRkKlE31MMvcwaFR9cduw | None | None | ... | True | False | 2026-05-20 20:13:55.494800+00:00 | 1 | 1 | 1 | 2 | 33 | 62 | 1 |
| 541718 | SgSa2b3L4b1yjIES0001 | scbasecount/2026-01-12/h5ad/GeneFull_Ex50pAS/H... | None | .h5ad | dataset | AnnData | 3748944 | 1aZuEAJSg+uoWb5nC1++FA | None | None | ... | True | False | 2026-05-20 20:13:55.494800+00:00 | 1 | 1 | 1 | 2 | 33 | 62 | 1 |
| 541703 | nitX6vDDuiWMvRga0001 | scbasecount/2026-01-12/h5ad/GeneFull_Ex50pAS/H... | None | .h5ad | dataset | AnnData | 3861565 | 7uv6ocCWHdOWYg1/wG9PXA | None | None | ... | True | False | 2026-05-20 20:13:55.494800+00:00 | 1 | 1 | 1 | 2 | 33 | 62 | 1 |
20 rows × 22 columns
Cache and load datasets into memory¶
Load the h5ads as a single AnnData by caching the datasets, concatenating them, and loading them into memory:
adata_concat = h5ads_brain[:5].load()
adata_concat
Show code cell output
/opt/hostedtoolcache/Python/3.12.13/x64/lib/python3.12/site-packages/lamindb/models/collection.py:94: UserWarning: Observation names are not unique. To make them unique, call `.obs_names_make_unique`.
concat_object = ad.concat(objects, join=join, label=label, keys=keys, **kwargs)
AnnData object with n_obs × n_vars = 34765 × 36601
obs: 'gene_count_Unique', 'umi_count_Unique', 'gene_count_UniqueAndMult-EM', 'umi_count_UniqueAndMult-EM', 'gene_count_UniqueAndMult-Uniform', 'umi_count_UniqueAndMult-Uniform', 'SRX_accession', 'cell_type', 'cell_ontology_term_id', 'artifact_uid'
layers: 'UniqueAndMult-EM', 'UniqueAndMult-Uniform', None (.X)
Open the sample metadata:
sample_meta = db.Artifact.get(
version_tag="2026-01-12",
key__endswith="sample_metadata.parquet",
projects=scbase,
organisms=organisms.human,
ulabels=feature_counts.genefull_ex50pas,
)
sample_meta_dataset = sample_meta.open()
sample_meta_dataset.schema
Show code cell output
entrez_id: int64
srx_accession: string
file_path: string
obs_count: int64
lib_prep: string
tech_10x: string
cell_prep: string
organism: string
tissue: string
tissue_ontology_term_id: string
disease: string
disease_ontology_term_id: string
perturbation: string
cell_line: string
antibody_derived_tag: string
czi_collection_id: string
czi_collection_name: string
single_disease_confidence: string
single_disease_confidence_reasoning: string
-- schema metadata --
pandas: '{"index_columns": [], "column_indexes": [], "columns": [{"name":' + 2527
Query the corresponding sample metadata:
filter_expr = pc.field("srx_accession").isin(
adata_concat.obs["SRX_accession"].astype(str)
)
df = sample_meta_dataset.scanner(filter=filter_expr).to_table().to_pandas()
Add the sample metadata to the AnnData object:
adata_concat.obs = adata_concat.obs.merge(
df, left_on="SRX_accession", right_on="srx_accession"
)
adata_concat
Show code cell output
/opt/hostedtoolcache/Python/3.12.13/x64/lib/python3.12/functools.py:912: ImplicitModificationWarning: Transforming to str index.
return dispatch(args[0].__class__)(*args, **kw)
AnnData object with n_obs × n_vars = 34765 × 36601
obs: 'gene_count_Unique', 'umi_count_Unique', 'gene_count_UniqueAndMult-EM', 'umi_count_UniqueAndMult-EM', 'gene_count_UniqueAndMult-Uniform', 'umi_count_UniqueAndMult-Uniform', 'SRX_accession', 'cell_type', 'cell_ontology_term_id', 'artifact_uid', 'entrez_id', 'srx_accession', 'file_path', 'obs_count', 'lib_prep', 'tech_10x', 'cell_prep', 'organism', 'tissue', 'tissue_ontology_term_id', 'disease', 'disease_ontology_term_id', 'perturbation', 'cell_line', 'antibody_derived_tag', 'czi_collection_id', 'czi_collection_name', 'single_disease_confidence', 'single_disease_confidence_reasoning'
layers: 'UniqueAndMult-EM', 'UniqueAndMult-Uniform', None (.X)
See the metadata in the AnnData:
adata_concat.obs.head()
Show code cell output
| gene_count_Unique | umi_count_Unique | gene_count_UniqueAndMult-EM | umi_count_UniqueAndMult-EM | gene_count_UniqueAndMult-Uniform | umi_count_UniqueAndMult-Uniform | SRX_accession | cell_type | cell_ontology_term_id | artifact_uid | ... | tissue_ontology_term_id | disease | disease_ontology_term_id | perturbation | cell_line | antibody_derived_tag | czi_collection_id | czi_collection_name | single_disease_confidence | single_disease_confidence_reasoning | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | 1 | 1.0 | 1 | 1.0 | 1 | 1.0 | SRX25506069 | qvSEhDQmKxucI3760000 | ... | UBERON:0000955 | glioblastoma | MONDO:0018177 | not mentioned | MGG23 | maybe | None | None | low | BioSample is silent (biosample_disclosed_disea... | ||
| 1 | 2 | 2.0 | 2 | 2.0 | 2 | 2.0 | SRX25506069 | qvSEhDQmKxucI3760000 | ... | UBERON:0000955 | glioblastoma | MONDO:0018177 | not mentioned | MGG23 | maybe | None | None | low | BioSample is silent (biosample_disclosed_disea... | ||
| 2 | 7 | 7.0 | 7 | 7.0 | 7 | 7.0 | SRX25506069 | qvSEhDQmKxucI3760000 | ... | UBERON:0000955 | glioblastoma | MONDO:0018177 | not mentioned | MGG23 | maybe | None | None | low | BioSample is silent (biosample_disclosed_disea... | ||
| 3 | 1 | 1.0 | 1 | 1.0 | 1 | 1.0 | SRX25506069 | qvSEhDQmKxucI3760000 | ... | UBERON:0000955 | glioblastoma | MONDO:0018177 | not mentioned | MGG23 | maybe | None | None | low | BioSample is silent (biosample_disclosed_disea... | ||
| 4 | 2 | 2.0 | 2 | 2.0 | 2 | 2.0 | SRX25506069 | qvSEhDQmKxucI3760000 | ... | UBERON:0000955 | glioblastoma | MONDO:0018177 | not mentioned | MGG23 | maybe | None | None | low | BioSample is silent (biosample_disclosed_disea... |
5 rows × 29 columns
Explore collections¶
This project has 135 collections of artifacts (27 organisms x 5 count features) for the latest version:
db.Collection.filter(version_tag="2026-01-12", projects=scbase).to_dataframe()
Show code cell output
! truncated query result to limit=20 Collection objects
| uid | key | description | hash | reference | reference_type | version_tag | is_latest | is_locked | created_at | branch_id | created_on_id | space_id | created_by_id | run_id | schema_id | meta_artifact_id | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| id | |||||||||||||||||
| 242 | Aioyo5zHXzPUkSuT0001 | scBaseCount/Velocyto/Bos_taurus | None | olJQs43iwGReP02Ig0bsMg | None | None | 2026-01-12 | True | False | 2026-05-21 11:26:48.999352+00:00 | 1 | 1 | 1 | 1 | 33 | None | None |
| 241 | gJrcdOm2sG7JUINS0001 | scBaseCount/GeneFull_ExonOverIntron/Bos_taurus | None | fHZY39j5Tl2HzuhH4JkbMA | None | None | 2026-01-12 | True | False | 2026-05-21 11:26:41.691858+00:00 | 1 | 1 | 1 | 1 | 33 | None | None |
| 240 | gY3xsMES4idjZb320001 | scBaseCount/GeneFull_Ex50pAS/Bos_taurus | None | Z-jbRmTLrqXa1OSnX0vXLg | None | None | 2026-01-12 | True | False | 2026-05-21 11:26:34.363847+00:00 | 1 | 1 | 1 | 1 | 33 | None | None |
| 239 | owfF1Bfuq660eiDp0001 | scBaseCount/GeneFull/Bos_taurus | None | z0dDiak_8xV-nqOAaOCMUQ | None | None | 2026-01-12 | True | False | 2026-05-21 11:26:27.041837+00:00 | 1 | 1 | 1 | 1 | 33 | None | None |
| 238 | ttGkPgXxLDO4sSXF0001 | scBaseCount/Gene/Bos_taurus | None | -FAJ3zwNRX34JZMFNWiGrQ | None | None | 2026-01-12 | True | False | 2026-05-21 11:26:19.701347+00:00 | 1 | 1 | 1 | 1 | 33 | None | None |
| 237 | HygGVUaaxxDSllWN0001 | scBaseCount/Velocyto/Macaca_mulatta | None | x4M52zpM7SnkM3_RB-hoKA | None | None | 2026-01-12 | True | False | 2026-05-21 11:26:12.030845+00:00 | 1 | 1 | 1 | 1 | 33 | None | None |
| 236 | OeaaN8NFmhu7ZvTb0001 | scBaseCount/GeneFull_ExonOverIntron/Macaca_mul... | None | iGbb7RJdVknw0eYMK5bvAw | None | None | 2026-01-12 | True | False | 2026-05-21 11:26:03.332097+00:00 | 1 | 1 | 1 | 1 | 33 | None | None |
| 235 | bI8Tk6DUPm5Q5pEo0001 | scBaseCount/GeneFull_Ex50pAS/Macaca_mulatta | None | d1nX4rRvA2SGveDKaSu_Gw | None | None | 2026-01-12 | True | False | 2026-05-21 11:25:54.516835+00:00 | 1 | 1 | 1 | 1 | 33 | None | None |
| 234 | WeEGa8CknImd804x0001 | scBaseCount/GeneFull/Macaca_mulatta | None | pfIlE2xynQVVDTiY3339xw | None | None | 2026-01-12 | True | False | 2026-05-21 11:25:45.021611+00:00 | 1 | 1 | 1 | 1 | 33 | None | None |
| 233 | TMcFueJifRSFVrSq0001 | scBaseCount/Gene/Macaca_mulatta | None | BQHkEvqCXlKfgcUJiz4Qsw | None | None | 2026-01-12 | True | False | 2026-05-21 11:25:36.100602+00:00 | 1 | 1 | 1 | 1 | 33 | None | None |
| 232 | lL7g9qDuJcrGwsHK0001 | scBaseCount/Velocyto/Oryza_sativa | None | slgnqMatWpHz2LtkWOxj2Q | None | None | 2026-01-12 | True | False | 2026-05-21 11:25:27.768084+00:00 | 1 | 1 | 1 | 1 | 33 | None | None |
| 231 | rForlsvLjM8zEgbO0001 | scBaseCount/GeneFull_ExonOverIntron/Oryza_sativa | None | P402N0vxF3-rKqal2u5S7g | None | None | 2026-01-12 | True | False | 2026-05-21 11:25:20.500858+00:00 | 1 | 1 | 1 | 1 | 33 | None | None |
| 230 | q4sDOLBVyJXVjqxB0001 | scBaseCount/GeneFull_Ex50pAS/Oryza_sativa | None | 10xmx4tPR2ocBQ6Im5aFgQ | None | None | 2026-01-12 | True | False | 2026-05-21 11:25:13.226342+00:00 | 1 | 1 | 1 | 1 | 33 | None | None |
| 229 | sxXttvFd0lJgnPXZ0001 | scBaseCount/GeneFull/Oryza_sativa | None | Zeap4qrZ2qYvHeMJDlNBXQ | None | None | 2026-01-12 | True | False | 2026-05-21 11:25:05.950101+00:00 | 1 | 1 | 1 | 1 | 33 | None | None |
| 228 | wXctL2347aWNGnf90001 | scBaseCount/Gene/Oryza_sativa | None | pBzfdxfssHrHdxyjXcIxCw | None | None | 2026-01-12 | True | False | 2026-05-21 11:24:58.672589+00:00 | 1 | 1 | 1 | 1 | 33 | None | None |
| 227 | DjyZcxk1MiRJjaAd0001 | scBaseCount/Velocyto/Sus_scrofa | None | EfB7Kcaod1Gx68mhFzibyg | None | None | 2026-01-12 | True | False | 2026-05-21 11:24:51.203095+00:00 | 1 | 1 | 1 | 1 | 33 | None | None |
| 226 | YM7WtFvA9S1b6c9K0001 | scBaseCount/GeneFull_ExonOverIntron/Sus_scrofa | None | tCGN-CzCc0J9_V5H7l7slw | None | None | 2026-01-12 | True | False | 2026-05-21 11:24:43.756605+00:00 | 1 | 1 | 1 | 1 | 33 | None | None |
| 225 | C0v0VS7zDQeeM5tx0001 | scBaseCount/GeneFull_Ex50pAS/Sus_scrofa | None | y1NZkFYL7u_vfMAAexxiiA | None | None | 2026-01-12 | True | False | 2026-05-21 11:24:36.254108+00:00 | 1 | 1 | 1 | 1 | 33 | None | None |
| 224 | dqrrbBzdgLqz7OJ20001 | scBaseCount/GeneFull/Sus_scrofa | None | YgvkzQWxjnXGOHpEr51onQ | None | None | 2026-01-12 | True | False | 2026-05-21 11:24:28.847843+00:00 | 1 | 1 | 1 | 1 | 33 | None | None |
| 223 | kcWyQbnFbSxQFCLL0001 | scBaseCount/Gene/Sus_scrofa | None | nq1vnK82prVTJQFhR7TNZg | None | None | 2026-01-12 | True | False | 2026-05-21 11:24:21.431099+00:00 | 1 | 1 | 1 | 1 | 33 | None | None |
Collections are immutable collections of artifacts, useful for model training or analytical workflows that need to rely on an immutable set rather than a mutable set of artifact that’s grouped by a folder or label annotation.
Show code cell content
assert db.bionty.CellLine.filter(artifacts__in=artifacts_tahoe).distinct().count() == 50
assert db.pertdb.Compound.filter(artifacts__in=artifacts_tahoe).distinct().count() == 380
assert (
db.pertdb.CompoundPerturbation.filter(artifacts__in=artifacts_tahoe)
.distinct()
.count()
== 1138
)