Curate datasets 
 ¶
¶
Curating a dataset means three things:
Validate that the dataset matches a desired schema.
If validation fails, standardize the dataset (e.g., by fixing typos, mapping synonyms) or update registries.
Annotate the dataset by linking it against metadata entities so that it becomes queryable.
In other guides, we’ve mostly covered annotation. In this guide we’ll curate common data structures focusing on validation and standardization.
Schema design patterns¶
A Schema is a specification that defines the expected structure, data types, and validation rules for a dataset.
Unlike pydantic.Model for dictionaries or pandera.Schema / pyarrow.lib.Schema for tables, a lamindb schema enables queries in a database and supports complex data structures.
Here is an FAQ that compares it with pydantic and pandera.
Schemas ensure data consistency by defining:
Which features exist in your dataset
What data types those features should have
What values are valid for categorical features
Which features are required vs optional
An exemplary schema that leverages Feature objects to define features:
schema = ln.Schema(
name="experiment_schema", # human-readable name
features=[ # required features
ln.Feature(name="cell_type", dtype=bt.CellType),
ln.Feature(name="treatment", dtype=str),
],
otype="DataFrame" # object type (DataFrame, AnnData, etc.)
)
Or for a composite data structure, like an AnnData:
schema = ln.Schema(
otype="AnnData",
slots={
"obs": cell_metadata_schema, # (pseudocode) cell annotations
"var.T": gene_id_schema # (pseudocode) gene-derived features
}
)
What are slots?
For composite data structures, you need to specify which component contains which schema, for example, to validate both cell metadata in .obs and gene metadata in .var within the same schema.
Each slot is a key like "obs" for AnnData observations, "rna:var" for MuData modalities, or "attrs:nested:key" for SpatialData annotations.
Before diving into curation, let’s understand the different schema approaches and when to use each one. Think of schemas as rules that define what valid data should look like.
Flexible schema¶
Use when: You want to validate those columns whose names match feature names in your Feature registry.
import lamindb as ln
schema = ln.Schema(name="valid_features", itype=ln.Feature).save()
Minimal required schema¶
Use when: You need certain columns but want flexibility for additional metadata.
import lamindb as ln
mitype = ln.Feature.get(name="mini_immuno", is_type=True)
schema = ln.Schema(
name="mini_immuno",
features=[
ln.Feature.get(name="perturbation", type=mitype),
ln.Feature.get(name="cell_type_by_model", type=mitype),
ln.Feature.get(name="assay_oid", type=mitype),
ln.Feature.get(name="donor", type=mitype),
ln.Feature.get(name="concentration", type=mitype),
ln.Feature.get(name="treatment_time_h", type=mitype),
],
flexible=True, # _additional_ columns in a dataframe are validated & annotated
).save()
Strict Schema¶
Use when: You need complete control over data structure and values.
# Only allows specified columns
schema = ln.Schema(
features=[...], # (pseudocode)
minimal_set=True, # whether all passed features are required
maximal_set=False # whether additional features are allowed
)
DataFrame¶
If you’re not connected to a database, create one:
lamin init --storage ./test-curate --modules bionty
Show code cell output
→ initialized lamindb: testuser1/test-curate
Let’s import lamindb and optionally track this run:
import lamindb as ln
ln.track()
Show code cell output
→ connected lamindb: testuser1/test-curate
→ created Transform('69VZX9cvy3Qw0000', key='curate.ipynb'), started new Run('0CWRFTiObfnYXQr4') at 2026-07-17 13:47:37 UTC
→ notebook imports: lamindb
• tip: to identify the notebook across renames, pass the uid: ln.track("69VZX9cvy3Qw")
We’ll be working with the mini immuno dataset:
df = ln.examples.datasets.mini_immuno.get_dataset1(
with_cell_type_synonym=True, with_cell_type_typo=True
)
df
Show code cell output
| ENSG00000153563 | ENSG00000010610 | ENSG00000170458 | perturbation | sample_note | cell_type_by_expert | cell_type_by_model | assay_oid | concentration | treatment_time_h | donor | donor_ethnicity | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| sample1 | 1 | 3 | 5 | DMSO | was ok | B-cell | B cell | EFO:0008913 | 0.1% | 24 | D0001 | [Chinese, Singaporean Chinese] |
| sample2 | 2 | 4 | 6 | IFNG | looks naah | CD8-pos alpha-beta T cell | T cell | EFO:0008913 | 200 nM | 24 | D0002 | [Chinese, Han Chinese] |
| sample3 | 3 | 5 | 7 | DMSO | pretty! 🤩 | CD8-pos alpha-beta T cell | T cell | EFO:0008913 | 0.1% | 6 | None | [Chinese] |
We’ll now be walking through a sequence of steps to curate it.
(1) Set up your registries¶
Before creating a schema, ensure your registries have the right features and labels:
import bionty as bt
import lamindb as ln
# define valid labels
perturbation_type = ln.Record(name="Perturbation", is_type=True).save()
ln.Record(name="DMSO", type=perturbation_type).save()
ln.Record(name="IFNG", type=perturbation_type).save()
bt.CellType.from_source(name="B cell").save()
bt.CellType.from_source(name="T cell").save()
# define valid features
mitype = ln.Feature(name="mini_immuno", is_type=True).save()
ln.Feature(name="perturbation", dtype=perturbation_type, type=mitype).save()
ln.Feature(name="cell_type_by_expert", dtype=bt.CellType, type=mitype).save()
ln.Feature(name="cell_type_by_model", dtype=bt.CellType, type=mitype).save()
ln.Feature(
name="assay_oid",
dtype=bt.ExperimentalFactor.ontology_id,
type=mitype,
).save()
ln.Feature(name="concentration", dtype=str, type=mitype).save()
ln.Feature(
name="treatment_time_h",
dtype="num",
coerce=True,
type=mitype,
).save()
ln.Feature(name="donor", dtype=str, nullable=True, type=mitype).save()
ln.Feature(name="donor_ethnicity", dtype=list[bt.Ethnicity], type=mitype).save()
(2) Create your schema¶
Let’s instantiate the flexible schema we discussed earlier (available in our examples module):
schema = ln.examples.datasets.mini_immuno.define_mini_immuno_schema_flexible()
schema.describe()
Show code cell output
Schema: mini_immuno ├── uid: SBzdwSbMS7K2iXke run: 0CWRFTi (curate.ipynb) │ itype: Feature otype: None │ hash: p82oI4eb-6vLJtt6BcTJIw ordered_set: False │ maximal_set: False minimal_set: True │ branch: main space: all │ created_at: 2026-07-17 13:47:42 UTC created_by: testuser1 └── Features (6) └── name dtype optional nullable coerce default_value perturbation Record[Perturbation] ✗ ✓ ✗ unset cell_type_by_model bionty.CellType ✗ ✓ ✗ unset assay_oid bionty.ExperimentalFactor.ontology_id ✗ ✓ ✗ unset donor str ✗ ✓ ✗ unset concentration str ✗ ✓ ✗ unset treatment_time_h num ✗ ✓ ✓ unset
(3) Validate the dataset¶
Shortcut
If you expect the validation to pass, you can directly ingest a validated artifact via:
artifact = ln.Artifact.from_dataframe(df, key="examples/my_curated_dataset.parquet", schema=schema).save()
If you want full control over the validation process with access to standardization helpers, you can instantiate a Curator object. Its validate() method validates that your dataset adheres to the criteria defined by the schema.
It identifies which values are already validated (exist in the registries) and which are potentially problematic (do not yet exist in our registries).
try:
curator = ln.curators.DataFrameCurator(df, schema)
curator.validate()
except ln.errors.ValidationError as error:
print(error)
Show code cell output
2 terms not validated in feature 'cell_type_by_expert': 'B-cell', 'CD8-pos alpha-beta T cell'
1 synonym found: "B-cell" → "B cell"
→ curate synonyms via: curator.cat.standardize("cell_type_by_expert")
for remaining terms:
→ fix typos, remove non-existent values, or create objects via:
objects = bionty.CellType.from_values(['B-cell', 'CD8-pos alpha-beta T cell'], field='name').save()
(4) Fix validation errors¶
Check the non-validated terms:
curator.cat.non_validated
Show code cell output
{'cell_type_by_expert': ['B-cell', 'CD8-pos alpha-beta T cell']}
For cell_type_by_expert, we see 2 terms are not validated.
First, let’s standardize the synonym “B-cell” as suggested:
curator.cat.standardize("cell_type_by_expert")
# now we have only one non-validated cell type left
curator.cat.non_validated
Show code cell output
{'cell_type_by_expert': ['CD8-pos alpha-beta T cell']}
For “CD8-pos alpha-beta T cell”, let’s find out which cell type in the public ontology might be the actual match:
# to check the correct spelling of categories, pass `public=True` to get a lookup object from public ontologies
# use `lookup = curator.cat.lookup()` to get a lookup object of existing records in your instance
lookup = curator.cat.lookup(public=True)
# here is an example for the "cell_type_by_expert" feature
cell_types = lookup["cell_type_by_expert"]
cell_types.cd8_positive_alpha_beta_t_cell
Show code cell output
CellType(ontology_id='CL:0000625', name='CD8-positive, alpha-beta T cell', definition='A T Cell Expressing An Alpha-Beta T Cell Receptor And The Cd8 Coreceptor.', synonyms='CD8-positive, alpha-beta T-cell|CD8-positive, alpha-beta T lymphocyte|CD8-positive, alpha-beta T-lymphocyte', parents=array(['CL:0000791'], dtype=object))
Now that we have a match, let’s fix it:
# fix the cell type name
df["cell_type_by_expert"] = df["cell_type_by_expert"].cat.rename_categories(
{"CD8-pos alpha-beta T cell": cell_types.cd8_positive_alpha_beta_t_cell.name}
)
Validation now passes:
curator.validate()
(5) Save the dataset¶
artifact = curator.save_artifact(key="examples/my_curated_dataset.parquet")
artifact.describe()
Show code cell output
Artifact: examples/my_curated_dataset.parquet (0000) ├── uid: PKjbaLSfcYTuy8TE0000 run: 0CWRFTi (curate.ipynb) │ kind: dataset otype: DataFrame │ hash: 8YNnOL_jbSCFewcYdATlMQ size: 10.1 KB │ branch: main space: all │ created_at: 2026-07-17 13:47:45 UTC created_by: testuser1 │ n_observations: 3 schema: mini_immuno ├── storage/path: /home/runner/work/lamindb/lamindb/docs/test-curate/.lamindb/PKjbaLSfcYTuy8TE0000.parquet ├── Dataset features │ └── columns (8) │ assay_oid bionty.ExperimentalFactor.ontology… EFO:0008913 │ cell_type_by_expert bionty.CellType B cell, CD8-positive, alpha-beta T cell │ cell_type_by_model bionty.CellType B cell, T cell │ concentration str │ donor str │ donor_ethnicity list[bionty.Ethnicity] ['Chinese', 'Singaporean Chinese', 'Ha… │ perturbation Record[Perturbation] DMSO, IFNG │ treatment_time_h num └── Labels └── .records Record DMSO, IFNG .cell_types bionty.CellType B cell, T cell, CD8-positive, alpha-be… .experimental_factors bionty.ExperimentalFactor single-cell RNA sequencing .ethnicities bionty.Ethnicity Chinese, Singaporean Chinese, Han Chin…
Common fixes¶
This section covers the most frequent curation issues and their solutions. Use this as a reference when validation fails.
Feature validation errors¶
Issue: “Column not in dataframe”
"column 'treatment' not in dataframe. Columns in dataframe: ['drug', 'timepoint', ...]"
Solutions:
# Solution 1: Rename columns to match schema
df = df.rename(columns={
'treatment': 'drug',
'time': 'timepoint',
...
})
# Solution 2: Create missing columns
df['treatment'] = 'unknown' # Add with default value (or define Feature.default_value)
# Solution 3: Modify schema to match your data
schema = ln.Schema(
features=[
ln.Feature.get(name="drug"), # Use actual column name
ln.Feature.get(name="timepoint"),
],
...
)
Value validation errors¶
Issue: “Terms not validated in feature ‘cell_type’”
2 terms not validated in feature 'cell_type': 'B-cell', 'CD8-pos alpha-beta T cell'
1 synonym found: "B-cell" → "B cell"
→ curate synonyms via: curator.cat.standardize("cell_type")
for remaining terms:
→ fix typos, remove non-existent values, or create objects via:
objects = bionty.CellType.from_values(['CD8-pos alpha-beta T cell'], field='name').save()
Solutions:
# Solution 1: Use automatic standardization if given hint (handles synonyms)
curator.cat.standardize('cell_type')
# Solution 2: Manual mapping for complex cases
value_mapping = {
'T-cells': 'T cell',
'B-cells': 'B cell',
}
df['cell_type'] = df['cell_type'].map(value_mapping).fillna(df['cell_type'])
# Solution 3: Use public ontology lookup for correct names
lookup = curator.cat.lookup(public=True)
cell_types = lookup["cell_type"]
df['cell_type'] = df['cell_type'].cat.rename_categories({
'CD8-pos T cell': cell_types.cd8_positive_alpha_beta_t_cell.name
})
# Solution 4: Create new legitimate objects
objects = bt.CellType.from_values(["my_new_cell_type"]).save()
Data type errors¶
Issue: “Expected categorical data, got object”
TypeError: Expected categorical data for cell_type, got object
Solutions:
# Solution 1: Convert to categorical
df['cell_type'] = df['cell_type'].astype('category')
# Solution 2: Use coercion in feature definition
ln.Feature(name="cell_type", dtype=bt.CellType, coerce=True).save()
Organism-specific ontology errors¶
Issue: “Terms not validated” for organism-specific ontologies like developmental stages
2 terms not validated in feature 'developmental_stage_ontology_id': 'MmusDv:0000142', 'MmusDv:0000022'
Solution: Specify organism-specific source in feature definition using cat_filters:
# When defining the schema, specify the organism-specific source
mouse_source = bt.Source.filter(
entity="bionty.DevelopmentalStage",
organism="mouse"
).one()
schema = ln.Schema(
features=[
ln.Feature(
name="developmental_stage_ontology_id",
dtype=bt.DevelopmentalStage.ontology_id,
cat_filters={"source": mouse_source} # Specify organism-specific source
)
],
...
)
This pattern applies to any ontology where the same registry serves multiple organisms (e.g., DevelopmentalStage, Phenotype, …).
External data validation¶
Since not all metadata is always stored within the dataset itself, it is also possible to validate external metadata. For instance, you might want to validate a separate metadata dictionary or JSON file against a schema before attaching it to your data.
import lamindb as ln
from datetime import date
df = ln.examples.datasets.mini_immuno.get_dataset1(otype="DataFrame")
temperature = ln.Feature(name="temperature", dtype=float).save()
date_of_study = ln.Feature(name="date_of_study", dtype=date).save()
external_schema = ln.Schema(features=[temperature, date_of_study]).save()
concentration = ln.Feature(name="concentration", dtype=str).save()
donor = ln.Feature(name="donor", dtype=str, nullable=True).save()
schema = ln.Schema(
features=[concentration, donor],
slots={"__external__": external_schema},
otype="DataFrame",
).save()
artifact = ln.Artifact.from_dataframe(
df,
key="examples/dataset1.parquet",
features={"temperature": 21.6, "date_of_study": date(2024, 10, 1)},
schema=schema,
).save()
artifact.describe()
python scripts/curate_dataframe_external_features.py
Show code cell output
→ connected lamindb: testuser1/test-curate
! no run & transform got linked, call `ln.track()` & re-run
→ returning artifact with same hash: Artifact(uid='PKjbaLSfcYTuy8TE0000', key='examples/m
y_curated_dataset.parquet', description=None, suffix='.parquet', kind='dataset', otype='DataFrame',
size=10354, hash='8YNnOL_jbSCFewcYdATlMQ', n_files=None, n_observations=3, extra_data=None, branch_i
d=1, created_on_id=1, space_id=1, storage_id=1, run_id=1, schema_id=1, created_by_id=1, created_at=2
026-07-17 13:47:45 UTC, is_locked=False, version_tag=None, is_latest=True); to track this artifact a
s an input, use: ln.Artifact.get()
! key examples/my_curated_dataset.parquet on existing artifact differs from passed key exam
ples/dataset1.parquet, keeping original key; update manually if needed or pass skip_hash_lookup=True
if you want to duplicate the artifact
→ loading artifact into memory for validation
Artifact: examples/my_curated_dataset.parquet (0000)
├──
uid: PKjbaLSfcYTuy8TE0000 run: 0CWRFTi (curate.ipynb0
m)
│ kind: dataset otype: DataFrame
│ hash: 8YNnOL_jbSCFewcYdATlMQ size: 10.1 KB
│ branch: main space: all
│ created_at: 2026-07-17 13:47:45 UTC created_by: testuser1
│
n_observations: 3 schema: NKWxmhA
├──
storage/path:
│ /home/runner/work/lamindb/lamindb/docs/test-curate
/.lamindb/PKjbaLSfcYTuy8TE
│ 0000.parquet
├── Dataset featu
res
│ └── columns (2) 1
m
│
concentration str
2
m│ donor str
├── External features
│ └── assay_oid
bionty.ExperimentalFac… EFO:0008913
│
cell_type_by_expe… bionty.CellType B cell, CD8-positive, alph…
│ cell_type_by_model bionty.CellType B cell, T ce
ll
│ donor_ethnicity list[bionty.Ethnicity]
0m ['Chinese', 'Singaporean C…
│ perturbation Record[Perturbati
on] DMSO, IFNG
│ date_of_study da
te 2024-10-01
│ temperature
float 21.6
└── 1;
93mLabels
└── .records Record
[0m DMSO, IFNG
.cell_types bio
nty.CellType B cell, T cell, CD8-positi…
.experimenta
l_fac… bionty.ExperimentalFac… single-cell RNA sequencing
0
m .ethnicities bionty.Ethnicity Chinese, Singapore
an Chine…
Union dtypes¶
Some metadata columns might validate against several registries. This script demonstrates how to configure a feature that accepts values from multiple sources, such as allowing either a gene symbol or an Ensembl ID.
import lamindb as ln
import pandas as pd
union_feature = ln.Feature(
name="mixed_feature",
dtype="cat[bionty.Tissue.ontology_id|bionty.CellType.ontology_id]",
).save()
df_mixed = pd.DataFrame({"mixed_feature": ["UBERON:0000178", "CL:0000540"]})
schema = ln.Schema(features=[union_feature], coerce=True).save()
curator = ln.curators.DataFrameCurator(df_mixed, schema)
curator.validate()
python scripts/curate_dataframe_union_features.py
Show code cell output
→ connected lamindb: testuser1/test-curate
! rather than passing a string 'cat[bionty.Tissue.ontology_id|bionty.CellType.ontology_id]'
to dtype, consider passing a Python object
AnnData¶
AnnData, like all other data structures that follow, is a composite structure that stores different arrays in different slots.
Allow a flexible schema¶
We can also allow a flexible schema for an AnnData and only require that it’s indexed with Ensembl gene IDs.
import lamindb as ln
ln.examples.datasets.mini_immuno.define_features_labels()
adata = ln.examples.datasets.mini_immuno.get_dataset1(otype="AnnData")
artifact = ln.Artifact.from_anndata(
adata,
key="examples/mini_immuno.h5ad",
schema="ensembl_gene_ids_and_valid_features_in_obs",
).save()
artifact.describe()
Let’s run the script.
python scripts/curate_anndata_flexible.py
Show code cell output
→ connected lamindb: testuser1/test-curate
→ returning record with same name: 'Perturbation'
→ returning record with same name: 'DMSO'
→ returning record with same name: 'IFNG'
→ returning feature with same name: 'mini_immuno'
→ returning feature with same name: 'perturbation'
→ returning feature with same name: 'cell_type_by_expert'
→ returning feature with same name: 'cell_type_by_model'
→ returning feature with same name: 'assay_oid'
→ returning feature with same name: 'concentration'
→ returning feature with same name: 'treatment_time_h'
→ returning feature with same name: 'donor'
→ returning feature with same name: 'donor_ethnicity'
! no run & transform got linked, call `ln.track()` & re-run
→ loading artifact into memory for validation
Artifact: examples/mini_immuno.h5ad (0000)
├── uid: j
Jm45E1p3M2ATSGo0000 run:
│ kin
d: dataset otype: AnnData
│
hash: FB3CeMjmg1ivN6HDy6… size: 30.9 KB
│ branch: main space: all
│ created_at: 2026-07-17 1… created_by: testuser1
│ n_observations: 3 schema: anndata_ensembl_gene_ids
_and_valid_featu…
├── storage/path:
│ /home/runner/work/la
mindb/lamindb/docs/test-curate/.lamindb/jJm45E1p3M2ATSGo
│ 0000.h5ad
├
── Dataset features
│ ├── obs (7)
│ │ assay_oid bionty.ExperimentalFac…
[0m EFO:0008913
│ │ cell_type_by_expe… bionty.CellTyp
e B cell, CD8-positive, alph…
│ │ cell_type_by_model 0
mbionty.CellType B cell, T cell
│ │ concentrat
ion str
│ 0
m│ donor str
│ │ perturbation Record[Perturbation] DMSO, I
FNG
│ │ treatment_time_h num
│ └── var.T (3 bionty.G…
[0m
│ CD14 num
│ CD4 num
2m
│ CD8A num
└── Labels
[2m└── .records Record DMSO, IFNG
.cell_types bionty.CellType 2
m B cell, T cell, CD8-positi…
.experimental_fac… biont
y.ExperimentalFac… single-cell RNA sequencing
Under the hood, this uses the following built-in schema (anndata_ensembl_gene_ids_and_valid_features_in_obs()):
import bionty as bt
import lamindb as ln
obs_schema = ln.examples.schemas.valid_features()
varT_schema = ln.Schema(
name="valid_ensembl_gene_ids", itype=bt.Gene.ensembl_gene_id
).save()
schema = ln.Schema(
name="anndata_ensembl_gene_ids_and_valid_features_in_obs",
otype="AnnData",
slots={"obs": obs_schema, "var.T": varT_schema},
).save()
This schema transposes the var DataFrame during curation, so that one validates and annotates the columns of var.T, i.e., [ENSG00000153563, ENSG00000010610, ENSG00000170458].
If one doesn’t transpose, one would annotate the columns of var, i.e., [gene_symbol, gene_type].
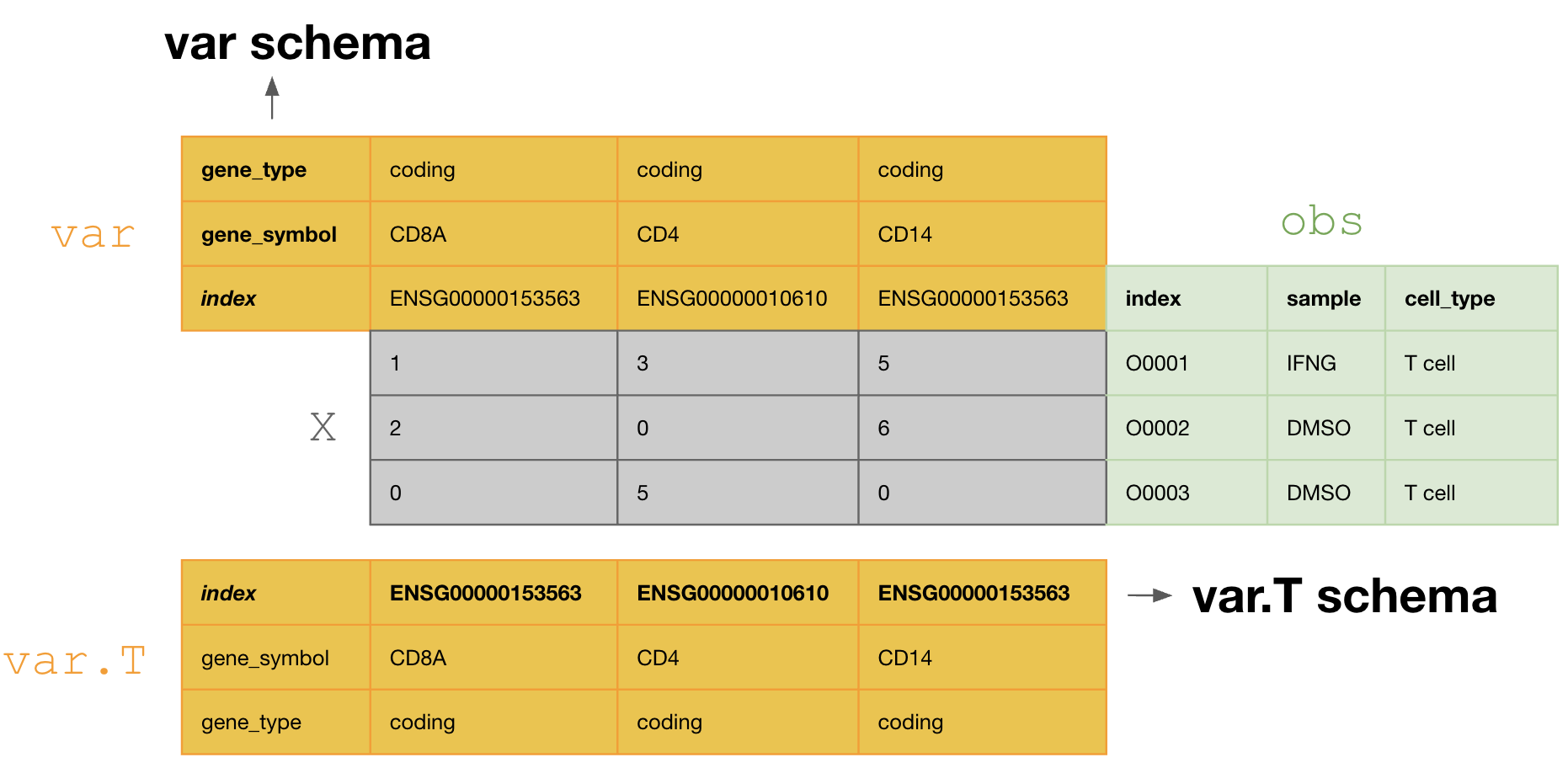
Fix validation issues¶
adata = ln.examples.datasets.mini_immuno.get_dataset1(
with_gene_typo=True, with_cell_type_typo=True, otype="AnnData"
)
adata
Show code cell output
AnnData object with n_obs × n_vars = 3 × 3
obs: 'perturbation', 'sample_note', 'cell_type_by_expert', 'cell_type_by_model', 'assay_oid', 'concentration', 'treatment_time_h', 'donor'
uns: 'temperature', 'experiment', 'date_of_study', 'study_note'
schema = ln.examples.schemas.anndata_ensembl_gene_ids_and_valid_features_in_obs()
schema.describe()
Show code cell output
Schema: anndata_ensembl_gene_ids_and_valid_features_in_obs ├── uid: 0000000000000002 run: │ itype: None otype: AnnData │ hash: aqGWHvyY49W_PHELUMiBMw ordered_set: False │ maximal_set: False minimal_set: True │ branch: main space: all │ created_at: 2026-07-17 13:47:57 UTC created_by: testuser1 ├── obs: valid_features │ └── uid: 0000000000000000 run: │ itype: Feature otype: None │ hash: kMi7B_N88uu-YnbTLDU-DA ordered_set: False │ maximal_set: False minimal_set: True │ branch: main space: all │ created_at: 2026-07-17 13:47:57 UTC created_by: testuser1 └── var.T: valid_ensembl_gene_ids ├── uid: 0000000000000001 run: │ itype: bionty.Gene.ensembl_gene_id otype: None │ hash: 1gocc_TJ1RU2bMwDRK-WUA ordered_set: False │ maximal_set: False minimal_set: True │ branch: main space: all │ created_at: 2026-07-17 13:47:57 UTC created_by: testuser1 └── bionty.Gene.ensembl_gene_id └── dtype: num
Check the slots of a schema:
schema.slots
Show code cell output
{'obs': Schema(uid='0000000000000000', is_type=False, name='valid_features', description=None, n_members=None, coerce=None, flexible=True, itype='Feature', otype=None, hash='kMi7B_N88uu-YnbTLDU-DA', minimal_set=True, ordered_set=False, maximal_set=False, branch_id=1, created_on_id=1, space_id=1, created_by_id=1, run_id=None, type_id=None, created_at=2026-07-17 13:47:57 UTC, is_locked=False),
'var.T': Schema(uid='0000000000000001', is_type=False, name='valid_ensembl_gene_ids', description=None, n_members=None, coerce=None, flexible=True, itype='bionty.Gene.ensembl_gene_id', otype=None, hash='1gocc_TJ1RU2bMwDRK-WUA', minimal_set=True, ordered_set=False, maximal_set=False, branch_id=1, created_on_id=1, space_id=1, created_by_id=1, run_id=None, type_id=None, created_at=2026-07-17 13:47:57 UTC, is_locked=False)}
curator = ln.curators.AnnDataCurator(adata, schema)
try:
curator.validate()
except ln.errors.ValidationError as error:
print(error)
Show code cell output
1 term not validated in feature 'cell_type_by_expert' in slot 'obs': 'CD8-pos alpha-beta T cell'
→ fix typos, remove non-existent values, or create objects via:
objects = bionty.CellType.from_values(['CD8-pos alpha-beta T cell'], field='name').save()
As above, we leverage a lookup object with valid cell types to find the correct name:
valid_cell_types = curator.slots["obs"].cat.lookup(public=True)["cell_type_by_expert"]
adata.obs["cell_type_by_expert"] = adata.obs[
"cell_type_by_expert"
].cat.rename_categories(
{"CD8-pos alpha-beta T cell": valid_cell_types.cd8_positive_alpha_beta_t_cell.name}
)
The validated AnnData can be subsequently saved as an Artifact:
adata.obs.columns
Show code cell output
Index(['perturbation', 'sample_note', 'cell_type_by_expert',
'cell_type_by_model', 'assay_oid', 'concentration', 'treatment_time_h',
'donor'],
dtype='object')
artifact = curator.save_artifact(key="examples/my_curated_anndata.h5ad")
Show code cell output
→ returning schema with same hash: Schema(uid='eAOyDwpwUZr6wnPS', is_type=False, name=None, description=None, n_members=7, coerce=None, flexible=False, itype='Feature', otype=None, hash='h1siSGrKKeoQCM9p7vGuzQ', minimal_set=True, ordered_set=False, maximal_set=False, branch_id=1, created_on_id=1, space_id=1, created_by_id=1, run_id=None, type_id=None, created_at=2026-07-17 13:48:01 UTC, is_locked=False)
! run was not set on Schema(uid='eAOyDwpwUZr6wnPS', is_type=False, name=None, description=None, n_members=7, coerce=None, flexible=False, itype='Feature', otype=None, hash='h1siSGrKKeoQCM9p7vGuzQ', minimal_set=True, ordered_set=False, maximal_set=False, branch_id=1, created_on_id=1, space_id=1, created_by_id=1, run_id=None, type_id=None, created_at=2026-07-17 13:48:01 UTC, is_locked=False), setting to current run
Access the schema for each slot:
artifact.features.slots
Show code cell output
{'obs': Schema(uid='eAOyDwpwUZr6wnPS', is_type=False, name=None, description=None, n_members=7, coerce=None, flexible=False, itype='Feature', otype=None, hash='h1siSGrKKeoQCM9p7vGuzQ', minimal_set=True, ordered_set=False, maximal_set=False, branch_id=1, created_on_id=1, space_id=1, created_by_id=1, run_id=None, type_id=None, created_at=2026-07-17 13:48:01 UTC, is_locked=False),
'var.T': Schema(uid='NVJoH1PF96eNRgCV', is_type=False, name=None, description=None, n_members=2, coerce=None, flexible=False, itype='bionty.Gene.ensembl_gene_id', otype=None, hash='UQ_O7OZzN0TyFt8pKzr_ZA', minimal_set=True, ordered_set=False, maximal_set=False, branch_id=1, created_on_id=1, space_id=1, created_by_id=1, run_id=1, type_id=None, created_at=2026-07-17 13:48:06 UTC, is_locked=False)}
The saved artifact has been annotated with validated features and labels:
artifact.describe()
Show code cell output
Artifact: examples/my_curated_anndata.h5ad (0000) ├── uid: qJ5RjdqVaFnvwLcz0000 run: 0CWRFTi (curate.ipynb) │ kind: dataset otype: AnnData │ hash: yeNWx0-dOGGkANQbocU4Sg size: 30.9 KB │ branch: main space: all │ created_at: 2026-07-17 13:48:06 UTC created_by: testuser1 │ n_observations: 3 schema: anndata_ensembl_gene_ids_and_valid_features_in_obs ├── storage/path: /home/runner/work/lamindb/lamindb/docs/test-curate/.lamindb/qJ5RjdqVaFnvwLcz0000.h5ad ├── Dataset features │ ├── obs (7) │ │ assay_oid bionty.ExperimentalFactor.ontology… EFO:0008913 │ │ cell_type_by_expert bionty.CellType B cell, CD8-positive, alpha-beta T cell │ │ cell_type_by_model bionty.CellType B cell, T cell │ │ concentration str │ │ donor str │ │ perturbation Record[Perturbation] DMSO, IFNG │ │ treatment_time_h num │ └── var.T (2 bionty.Gene.ensembl… │ CD4 num │ CD8A num └── Labels └── .records Record DMSO, IFNG .cell_types bionty.CellType B cell, T cell, CD8-positive, alpha-be… .experimental_factors bionty.ExperimentalFactor single-cell RNA sequencing
Unstructured dictionaries¶
Most data structures support unstructured metadata stored as dictionaries:
Pandas DataFrames:
.attrsAnnData:
.unsMuData:
.unsandmodality:unsSpatialData:
.attrs
These dictionaries can be nested, and sometimes you want to define a schema
specifically for a dictionary inside the dictionary. Here, we look at the case where
we want to validate a key within the .uns slot of an AnnData to ensure that they contain the "experiment" and "temperature" keys:
{
"study_metadata": {"experiment": "Experiment 1", "temperature": 21.2}
}
Here, we demonstrate how to curate such metadata for AnnData:
import lamindb as ln
from define_schema_df_metadata import study_metadata_schema
anndata_uns_schema = ln.Schema(
name="anndata_nested_study_metadata",
otype="AnnData",
slots={
"uns:study_metadata": study_metadata_schema,
},
).save()
python scripts/define_schema_anndata_uns.py
Show code cell output
→ connected lamindb: testuser1/test-curate
→ returning feature with same name: 'temperature'
import lamindb as ln
ln.examples.datasets.mini_immuno.define_features_labels()
adata = ln.examples.datasets.mini_immuno.get_dataset1(otype="AnnData")
adata.uns["study_metadata"] = adata.uns.copy()
schema = ln.Schema.get(name="anndata_nested_study_metadata")
artifact = ln.Artifact.from_anndata(
adata, schema=schema, key="examples/mini_immuno_uns.h5ad"
)
artifact.describe()
python scripts/curate_anndata_uns.py
Show code cell output
→ connected lamindb: testuser1/test-curate
→ returning record with same name: 'Perturbation'
→ returning record with same name: 'DMSO'
→ returning record with same name: 'IFNG'
→ returning feature with same name: 'mini_immuno'
→ returning feature with same name: 'perturbation'
→ returning feature with same name: 'cell_type_by_expert'
→ returning feature with same name: 'cell_type_by_model'
→ returning feature with same name: 'assay_oid'
→ returning feature with same name: 'concentration'
→ returning feature with same name: 'treatment_time_h'
→ returning feature with same name: 'donor'
→ returning feature with same name: 'donor_ethnicity'
! no run & transform got linked, call `ln.track()` & re-run
→ loading artifact into memory for validation
Artifact: examples/mini_immuno_uns.h5ad (0000)
├── uid:
[0mKEg7SwAXz5Cbz30f0000 run:
│ 2
mkind: dataset otype: AnnData
│
hash: fiY_0ATnu06_F1Kw-n1Vnw size: 33.3 KB
2
m│ branch: main space: all
│ created_at: <django.db.models.expressions.DatabaseDefau… created_by
: testu…
│ n_observations: 3 s
chema: anndata_n…
└── storage/path:
/home/runner/work/
lamindb/lamindb/docs/test-curate/.lamindb/KEg7SwAXz5Cbz30f
0000.h5ad
MuData¶
MuData objects contain multiple modalities, each with its own structure. The following script shows how to define and validate schemas across different modalities (e.g., RNA and ATAC) simultaneously.
import lamindb as ln
import bionty as bt
from docs.scripts.define_schema_df_metadata import study_metadata_schema
# define labels
perturbation = ln.Record(name="Perturbation", is_type=True).save()
ln.Record(name="Perturbed", type=perturbation).save()
ln.Record(name="NT", type=perturbation).save()
replicate = ln.Record(name="Replicate", is_type=True).save()
ln.Record(name="rep1", type=replicate).save()
ln.Record(name="rep2", type=replicate).save()
ln.Record(name="rep3", type=replicate).save()
# define the global obs schema
obs_schema = ln.Schema(
name="mudata_papalexi21_subset_obs_schema",
features=[
ln.Feature(name="perturbation", dtype=perturbation).save(),
ln.Feature(name="replicate", dtype=replicate).save(),
],
).save()
# define the ['rna'].obs schema
obs_schema_rna = ln.Schema(
name="mudata_papalexi21_subset_rna_obs_schema",
features=[
ln.Feature(name="nCount_RNA", dtype=int).save(),
ln.Feature(name="nFeature_RNA", dtype=int).save(),
ln.Feature(name="percent.mito", dtype=float).save(),
],
).save()
# define the ['hto'].obs schema
obs_schema_hto = ln.Schema(
name="mudata_papalexi21_subset_hto_obs_schema",
features=[
ln.Feature(name="nCount_HTO", dtype=int).save(),
ln.Feature(name="nFeature_HTO", dtype=int).save(),
ln.Feature(name="technique", dtype=bt.ExperimentalFactor).save(),
],
).save()
# define ['rna'].var schema
var_schema_rna = ln.Schema(
name="mudata_papalexi21_subset_rna_var_schema",
itype=bt.Gene.symbol,
dtype=float,
).save()
# define composite schema
mudata_schema = ln.Schema(
name="mudata_papalexi21_subset_mudata_schema",
otype="MuData",
slots={
"obs": obs_schema,
"rna:obs": obs_schema_rna,
"hto:obs": obs_schema_hto,
"rna:var": var_schema_rna,
"uns:study_metadata": study_metadata_schema,
},
).save()
# curate a MuData
mdata = ln.examples.datasets.mudata_papalexi21_subset(with_uns=True)
bt.settings.organism = "human" # set the organism to map gene symbols
curator = ln.curators.MuDataCurator(mdata, mudata_schema)
artifact = curator.save_artifact(key="examples/mudata_papalexi21_subset.h5mu")
assert artifact.schema == mudata_schema
python scripts/curate_mudata.py
Show code cell output
→ connected lamindb: testuser1/test-curate
→ returning feature with same name: 'temperature'
→ returning feature with same name: 'experiment'
→ returning schema with same hash: Schema(uid='B7AQbfEHF8aoSrXP', is_type=False, name='St
udy metadata schema', description=None, n_members=2, coerce=None, flexible=False, itype='Feature', o
type=None, hash='GtVneV_4i2t4svm6dKdt3g', minimal_set=True, ordered_set=False, maximal_set=False, br
anch_id=1, created_on_id=1, space_id=1, created_by_id=1, run_id=None, type_id=None, created_at=2026-
07-17 13:48:08 UTC, is_locked=False)
→ returning record with same name: 'Perturbation'
! you are trying to create a record with name='nFeature_HTO' but a record with similar name
exists: 'nFeature_RNA'. Did you mean to load it?
/opt/hostedtoolcache/Python/3.13.14/x64/lib/python3.13/site-packages/mudata/_core/mudata.py:1841: Se
ttingWithCopyWarning:
A value is trying to be set on a copy of a slice from a DataFrame
See the ca
veats in the documentation: https://pandas.pydata.org/pandas-docs/stable/user_guide/indexing.html#re
turning-a-view-versus-a-copy
mod_df.rename(
! auto-transposed `var` for backward compat, please indicate transposition in the schema de
finition by calling out `.T`: slots={'var.T': itype=bt.Gene.ensembl_gene_id}
! no run & transform got linked, call `ln.track()` & re-run
→ returning schema with same hash: Schema(uid='t2P4gGaP2QPxw2wP', is_type=False, name='mu
data_papalexi21_subset_obs_schema', description=None, n_members=2, coerce=None, flexible=False, ityp
e='Feature', otype=None, hash='f8SQdOl4RlRX7vqjaexOPw', minimal_set=True, ordered_set=False, maximal
_set=False, branch_id=1, created_on_id=1, space_id=1, created_by_id=1, run_id=None, type_id=None, cr
eated_at=2026-07-17 13:48:14 UTC, is_locked=False)
→ returning schema with same hash: Schema(uid='jikYQijVXTRZFg09', is_type=False, name='mu
data_papalexi21_subset_rna_obs_schema', description=None, n_members=3, coerce=None, flexible=False,
itype='Feature', otype=None, hash='zUHvvGaxe8yedmX8bX5D3A', minimal_set=True, ordered_set=False, max
imal_set=False, branch_id=1, created_on_id=1, space_id=1, created_by_id=1, run_id=None, type_id=None
, created_at=2026-07-17 13:48:14 UTC, is_locked=False)
→ returning schema with same hash: Schema(uid='NnXz6OEzhtYlafc2', is_type=False, name='mu
data_papalexi21_subset_hto_obs_schema', description=None, n_members=3, coerce=None, flexible=False,
itype='Feature', otype=None, hash='iadFAam2ZLcKNVDnm7VV_Q', minimal_set=True, ordered_set=False, max
imal_set=False, branch_id=1, created_on_id=1, space_id=1, created_by_id=1, run_id=None, type_id=None
, created_at=2026-07-17 13:48:14 UTC, is_locked=False)
→ returning schema with same hash: Schema(uid='B7AQbfEHF8aoSrXP', is_type=False, name='St
udy metadata schema', description=None, n_members=2, coerce=None, flexible=False, itype='Feature', o
type=None, hash='GtVneV_4i2t4svm6dKdt3g', minimal_set=True, ordered_set=False, maximal_set=False, br
anch_id=1, created_on_id=1, space_id=1, created_by_id=1, run_id=None, type_id=None, created_at=2026-
07-17 13:48:08 UTC, is_locked=False)
SpatialData¶
For SpatialData, we need to validate annotations nested deep within the .attrs dictionary.
This script illustrates how to target and curate these nested metadata fields.
import lamindb as ln
import bionty as bt
# a very comprehensive schema for different slots of a SpatialData object
# define or query features
bio_dict = ln.Feature(name="bio", dtype=dict).save()
tech_dict = ln.Feature(name="tech", dtype=dict).save()
disease = ln.Feature(name="disease", dtype=bt.Disease, coerce=True).save()
developmental_stage = ln.Feature(
name="developmental_stage",
dtype=bt.DevelopmentalStage,
coerce=True,
).save()
assay = ln.Feature(name="assay", dtype=bt.ExperimentalFactor, coerce=True).save()
sample_region = ln.Feature(name="sample_region", dtype=str).save()
analysis = ln.Feature(name="analysis", dtype=str).save()
# define or query schema components
attrs_schema = ln.Schema([bio_dict, tech_dict]).save()
sample_schema = ln.Schema([disease, developmental_stage]).save()
tech_schema = ln.Schema([assay]).save()
obs_schema = ln.Schema([sample_region]).save()
uns_schema = ln.Schema([analysis]).save()
# enforces only registered Ensembl Gene IDs pass validation (maximal_set=True)
varT_schema = ln.Schema(itype=bt.Gene.ensembl_gene_id, maximal_set=True).save()
# compose the SpatialData schema
sdata_schema = ln.Schema(
name="spatialdata_blobs_schema",
otype="SpatialData",
slots={
"attrs:bio": sample_schema,
"attrs:tech": tech_schema,
"attrs": attrs_schema,
"tables:table:obs": obs_schema,
"tables:table:var.T": varT_schema,
},
).save()
python scripts/define_schema_spatialdata.py
Show code cell output
→ connected lamindb: testuser1/test-curate
! you are trying to create a record with name='tech' but a record with similar name exists:
'technique'. Did you mean to load it?
import lamindb as ln
spatialdata = ln.examples.datasets.spatialdata_blobs()
sdata_schema = ln.Schema.get(name="spatialdata_blobs_schema")
curator = ln.curators.SpatialDataCurator(spatialdata, sdata_schema)
try:
curator.validate()
except ln.errors.ValidationError:
pass
spatialdata.tables["table"].var.drop(index="ENSG00000999999", inplace=True)
# validate again (must pass now) and save artifact
artifact = ln.Artifact.from_spatialdata(
spatialdata, key="examples/spatialdata1.zarr", schema=sdata_schema
).save()
artifact.describe()
python scripts/curate_spatialdata.py
Show code cell output
→ connected lamindb: testuser1/test-curate
/opt/hostedtoolcache/Python/3.13.14/x64/lib/python3.13/site-packages/spatialdata/_core/query/relatio
nal_query.py:531: FutureWarning: functools.partial will be a method descriptor in future Python vers
ions; wrap it in enum.member() if you want to preserve the old behavior
left = partial(_left_join_
spatialelement_table)
/opt/hostedtoolcache/Python/3.13.14/x64/lib/python3.13/site-packages/spatialda
ta/_core/query/relational_query.py:532: FutureWarning: functools.partial will be a method descriptor
in future Python versions; wrap it in enum.member() if you want to preserve the old behavior
left
_exclusive = partial(_left_exclusive_join_spatialelement_table)
/opt/hostedtoolcache/Python/3.13.14/
x64/lib/python3.13/site-packages/spatialdata/_core/query/relational_query.py:533: FutureWarning: fun
ctools.partial will be a method descriptor in future Python versions; wrap it in enum.member() if yo
u want to preserve the old behavior
inner = partial(_inner_join_spatialelement_table)
/opt/hostedt
oolcache/Python/3.13.14/x64/lib/python3.13/site-packages/spatialdata/_core/query/relational_query.py
:534: FutureWarning: functools.partial will be a method descriptor in future Python versions; wrap i
t in enum.member() if you want to preserve the old behavior
right = partial(_right_join_spatialele
ment_table)
/opt/hostedtoolcache/Python/3.13.14/x64/lib/python3.13/site-packages/spatialdata/_core/q
uery/relational_query.py:535: FutureWarning: functools.partial will be a method descriptor in future
Python versions; wrap it in enum.member() if you want to preserve the old behavior
right_exclusiv
e = partial(_right_exclusive_join_spatialelement_table)
! no run & transform got linked, call `ln.track()` & re-run
/opt/hostedtoolcache/Python/3.13.14/x64/lib/python3.13/site-packages/ome_zarr/writer.py:319: FutureW
arning: Passing storage-related arguments via **kwargs is deprecated. Please use the 'zarr_store_kwa
rgs' parameter instead. **kwargs will be removed in a future version.
da_delayed = da.to_zarr(
→ loading artifact into memory for validation
→ returning schema with same hash: Schema(uid='Y1C0Tl47ZgZ4IBk3', is_type=False, name=Non
e, description=None, n_members=2, coerce=None, flexible=False, itype='Feature', otype=None, hash='qE
oipp-h9Opz83ummH7P5w', minimal_set=True, ordered_set=False, maximal_set=False, branch_id=1, created_
on_id=1, space_id=1, created_by_id=1, run_id=None, type_id=None, created_at=2026-07-17 13:48:23 UTC,
is_locked=False)
→ returning schema with same hash: Schema(uid='oncNkVdEI5Gr6PbL', is_type=False, name=Non
e, description=None, n_members=1, coerce=None, flexible=False, itype='Feature', otype=None, hash='bU
zRC2B9STzmFnPbCtKY-Q', minimal_set=True, ordered_set=False, maximal_set=False, branch_id=1, created_
on_id=1, space_id=1, created_by_id=1, run_id=None, type_id=None, created_at=2026-07-17 13:48:23 UTC,
is_locked=False)
→ returning schema with same hash: Schema(uid='GrvblgRz0WqFjcwT', is_type=False, name=Non
e, description=None, n_members=2, coerce=None, flexible=False, itype='Feature', otype=None, hash='OW
3iaaOg_ef17QFciKhiSg', minimal_set=True, ordered_set=False, maximal_set=False, branch_id=1, created_
on_id=1, space_id=1, created_by_id=1, run_id=None, type_id=None, created_at=2026-07-17 13:48:23 UTC,
is_locked=False)
→ returning schema with same hash: Schema(uid='fNiJnftIatqx3Url', is_type=False, name=Non
e, description=None, n_members=1, coerce=None, flexible=False, itype='Feature', otype=None, hash='es
Eg72VwvLDJtA3S9C8Jxg', minimal_set=True, ordered_set=False, maximal_set=False, branch_id=1, created_
on_id=1, space_id=1, created_by_id=1, run_id=None, type_id=None, created_at=2026-07-17 13:48:23 UTC,
is_locked=False)
Artifact: examples/spatialdata1.zarr (0000)
├── uid:
XdrWp2RuIF37nApv0000 run:
│ kind: 0
mdataset otype: SpatialData
│ hash: 0
mlEwEJdWjpyiAPCkUuS-cfg size: 12.4 MB
│ branch:
[0mmain space: all
│ created_a
t: 2026-07-17 13:48:40 UTC created_by: testuser1
│ n_files:
79 schema: spatialdata_blobs_schema
├── stora
ge/path:
│ /home/runner/work/lamindb/lamindb/docs/test-curate/.lam
indb/XdrWp2RuIF37nApv
│ .zarr
├── Dataset features
│
0m├── attrs:bio (2)
│ │ developmental_sta
… bionty.DevelopmentalSt… adult stage
│ │
disease bionty.Disease Alzheimer disease
│ ├── attrs:tech (1)
│ │ assay
bionty.ExperimentalFac… Visium Spatial Gene Expres…
│ ├── attrs (2)
│ │ bio
dict
│
│ tech dict
│ ├── tables:table:obs …
│ │ sample_re
gion str
│
0m└── tables:table:var.… 0
m
│ BRAF num
│ BRCA2
2m num
└── 1;93
mLabels
└── .diseases bionty.Disease 0
m Alzheimer disease
.experimental_fac… bio
nty.ExperimentalFac… Visium Spatial Gene Expres…
.developme
ntal_st… bionty.DevelopmentalSt… adult stage
TiledbsomaExperiment¶
TileDB-SOMA experiments store large-scale single-cell data on disk.
Here we show how to validate the obs and var dataframes of a SOMA experiment without loading the entire dataset into memory.
import lamindb as ln
import bionty as bt
import tiledbsoma as soma
import tiledbsoma.io
adata = ln.examples.datasets.mini_immuno.get_dataset1(otype="AnnData")
tiledbsoma.io.from_anndata("small_dataset.tiledbsoma", adata, measurement_name="RNA")
obs_schema = ln.Schema(
name="soma_obs_schema",
features=[
ln.Feature(name="cell_type_by_expert", dtype=bt.CellType).save(),
ln.Feature(name="cell_type_by_model", dtype=bt.CellType).save(),
],
).save()
var_schema = ln.Schema(
name="soma_var_schema",
features=[
ln.Feature(name="var_id", dtype=bt.Gene.ensembl_gene_id).save(),
],
coerce=True,
).save()
soma_schema = ln.Schema(
name="soma_experiment_schema",
otype="tiledbsoma",
slots={
"obs": obs_schema,
"ms:RNA.T": var_schema,
},
).save()
with soma.Experiment.open("small_dataset.tiledbsoma") as experiment:
curator = ln.curators.TiledbsomaExperimentCurator(experiment, soma_schema)
curator.validate()
artifact = curator.save_artifact(
key="examples/soma_experiment.tiledbsoma",
description="SOMA experiment with schema validation",
)
assert artifact.schema == soma_schema
artifact.describe()
python scripts/curate_soma_experiment.py
Show code cell output
→ connected lamindb: testuser1/test-curate
! no run & transform got linked, call `ln.track()` & re-run
→ returning schema with same hash: Schema(uid='Yooe5TSNT5GfjTZD', is_type=False, name='so
ma_obs_schema', description=None, n_members=2, coerce=None, flexible=False, itype='Feature', otype=N
one, hash='xuL-553axEBJnN0A0myzAg', minimal_set=True, ordered_set=False, maximal_set=False, branch_i
d=1, created_on_id=1, space_id=1, created_by_id=1, run_id=None, type_id=None, created_at=2026-07-17
13:48:45 UTC, is_locked=False)
→ returning schema with same hash: Schema(uid='WMgWXr5RNjCwB5Vo', is_type=False, name='so
ma_var_schema', description=None, n_members=1, coerce=True, flexible=False, itype='Feature', otype=N
one, hash='qs4bqU1L16c001HXWTho2g', minimal_set=True, ordered_set=False, maximal_set=False, branch_i
d=1, created_on_id=1, space_id=1, created_by_id=1, run_id=None, type_id=None, created_at=2026-07-17
13:48:45 UTC, is_locked=False)
Artifact: examples/soma_experiment.tiledbsoma (0000)
| description:
[0mSOMA experiment with schema validation
├── uid: NZekFuwF6bwxvUTy0000
run:
│ kind: dataset ot
ype: tiledbsoma
│ hash: X5zlY_cG0ZWKjh5CDRSIlA size: 23.
9 KB
│ branch: main space: all
│ created_at: 2026-07-17 13:48:46 UTC created_by: testuser1
│
n_observations: 3 n_files: 68
│ sc
hema: soma_experiment_schema
├── storage/path: 0
m
│ /home/runner/work/lamindb/lamindb/docs/test-curate/.lamindb/NZekFuw
F6bwxvUTy
│ .tiledbsoma
├── Dataset features
│ ├
── obs (2)
0m
│ │ cell_type_by_expe… 2
m bionty.CellType B cell, CD8-positive, alph…
│ │ cel
l_type_by_model bionty.CellType B cell, T cell
│ └── ms:RNA.T (1)
│ var_id
bionty.Gene.ensembl_ge… ENSG00000010610, ENSG00000…
└──
Labels
└── .genes bionty.Gene
CD8A, CD4, CD14
.cell_types
0mbionty.CellType B cell, T cell, CD8-positi…
Other data structures¶
If you have other data structures, read: How do I validate & annotate arbitrary data structures? .
rm -rf ./test-curate
rm -rf ./small_dataset.tiledbsoma
lamin delete --force test-curate