Introduction 




 ¶
¶
LaminDB is an open-source data management tool that makes it easy to query, trace & govern datasets across diverse storage formats and locations. It gives you context through annotations, memory through lineage, and governance through branching and versioning. It uses a scalable lakehouse architecture that integrates files, tables, arrays, ontologies, and notes. It supports biological data through modules for bio-formats and registries by the creators of Scanpy.
Why?
Untraceable results cannot be trusted, especially in the age of agents.
Without effective access to multimodal data, models burn tokens or fail entirely.
We want to govern changes to data like we govern changes to code with git.

How?
lineage → trace results across agent sessions, notebooks, scripts & workflows
lakehouse → query across many datasets, manage tables & arrays schema-based and ACID
LIMS & ELN → unified schema-based records management with support for ontologies & notes
FAIR datasets → validate & annotate files,
DataFrame,AnnData,SpatialData,zarr, …governance → manage changes via branching & by versioning data + code
Architecture?
zero lock-in → uses open standards (metadata in SQLite/Postgres, data in
parquet,zarr, etc.)scalable → hit storage & database directly through your
pydataor R stack, no REST API involvedsimple →
pip install lamindborinstall.packages('laminr')- no Docker required, no separate backendunified → federate data across storage locations (local, S3, GCP, …) in any database
distributed → federate data zero-copy & lineage-aware across databases
reproducible → track agent traces, source code & compute environments
idempotent → re-run logic without worries about duplications or overwrites
integrations → bio ontologies git, nextflow, vitessce, redun, and more
extensible → create custom plug-ins based on the Django ORM, the basis for LaminDB’s registries
Who?
Scientists and engineers at leading research institutions and biotech companies, including:
Industry → Pfizer, Altos Labs, Ensocell Therapeutics, …
Academia & Research → scverse, DZNE (National Research Center for Neuro-Degenerative Diseases), Helmholtz Munich (National Research Center for Environmental Health), …
Research Hospitals → Global Immunological Swarm Learning Network: Harvard, MIT, Stanford, ETH Zürich, Charité, U Bonn, Mount Sinai, …
From personal research projects to pharma-scale deployments managing petabytes of data across:
entities |
OOMs |
|---|---|
observations & datasets |
10¹² & 10⁶ |
runs & transforms |
10⁹ & 10⁵ |
proteins & genes |
10⁹ & 10⁶ |
biosamples & species |
10⁵ & 10² |
… |
… |
UI, permissions, audit logs?
LaminHub is a collaboration hub built on LaminDB similar to how GitHub is built on git.
Platform features:
infra-as-code → manage many distributed storage locations & databases
permissions → role-based, fine-grained access management for users & teams
audit logs → full traceability for compliance
single sign-on → connect Okta, Ping, and other providers
secure → SOC2 certified, monitoring ISO27001 & HIPAA compliance
Architecture features:
zero lock-in → the open-source core ensures data remains yours & accessible even if you cancel LaminHub
permissions on the Postgres & storage layer → no need for an intermediate web service
accredited TRE (Trusted Research Environment) → manage sensitive data from e.g. the UK Biobank
auto-generated REST API → optional REST interface for JS-based web applications
UI features:
lineage → interactive graphs for datasets, notebooks & pipelines
catalog → browse, search & query your lakehouse
notebooks, workflows, runs → visualize & launch executions
versioning → manage data & code revisions
LIMS & ELN → records, sheets & markdown notes integrated with ontologies
schemas & labels → validate & monitor data distributions
simple dashboarding → auto-generate data summaries
Give it a try by exploring public omics datasets at lamin.ai/explore. It’s free and no account is required.
LaminHub is a SaaS product. For private data & commercial usage, see: lamin.ai/pricing.
Quickstart¶
To install the Python package with recommended dependencies, use:
pip install lamindb
Install with minimal dependencies.
The lamindb package adds data-science related dependencies through the [full] extra, see here.
For a minimal install of the lamindb namespace, use:
pip install lamindb-core
Agent? The lamindb skill ships with the lamindb package at .agents/skills/. Pure markdown docs ship with the package in docs/ and are available from the web via llms.txt.
Query databases & datasets¶
You can browse public databases at lamin.ai/explore. To access laminlabs/cellxgene, run:
import lamindb as ln
db = ln.DB("laminlabs/cellxgene") # a database object for queries
df = db.Artifact.to_dataframe() # a dataframe listing datasets & models
library(laminr)
ln <- import_module("lamindb")
db <- ln$DB("laminlabs/cellxgene") # a database object for queries
df <- db$Artifact$to_dataframe() # a dataframe listing datasets & models
To get a specific dataset, run:
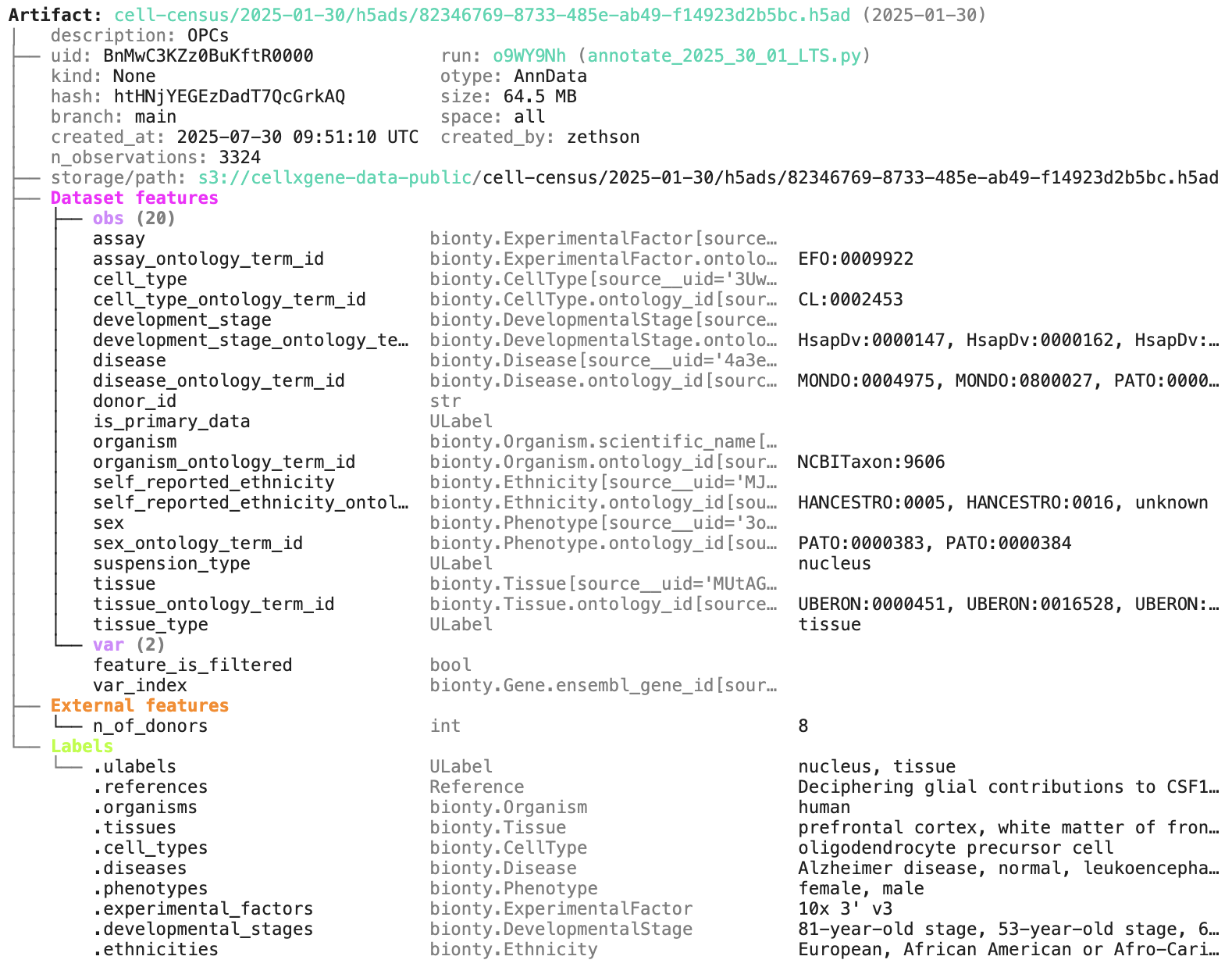
artifact = db.Artifact.get("BnMwC3KZz0BuKftR") # a metadata object for a dataset
artifact.describe() # describe the context of the dataset
artifact <- db$Artifact$get("BnMwC3KZz0BuKftR") # a metadata object for a dataset
artifact$describe() # describe the context of the dataset
See the output.
Access the content of the dataset via:
local_path = artifact.cache() # return a local path from a cache
adata = artifact.load() # load object into memory
accessor = artifact.open() # return a streaming accessor
local_path <- artifact$cache() # return a local path from a cache
adata <- artifact$load() # load object into memory
accessor <- artifact$open() # return a streaming accessor
For broader queries of cellxgene, see docs.lamin.ai/cellxgene.
Save files & folders¶
You can create a LaminDB instance at lamin.ai and invite collaborators. To connect to an existing instance, run:
lamin login
lamin connect account/name # tip: add flag `--here` to scope to current directory
Or init a new instance instead (no login required).
lamin init --storage ./quickstart-data --modules bionty
On the terminal and in a Python session, LaminDB will now auto-connect. For more configuration, see docs.lamin.ai/setup.
To save a file or folder via the API:
import lamindb as ln
# → connected lamindb: account/instance
open("sample.fasta", "w").write(">seq1\nACGT\n") # create dataset
ln.Artifact("sample.fasta", key="sample.fasta").save() # save dataset
library(laminr)
ln <- import_module("lamindb")
# → connected lamindb: account/instance
writeLines(">seq1\nACGT\n", "sample.fasta") # create dataset
ln$Artifact("sample.fasta", key = "sample.fasta")$save() # save dataset
To save a file or folder via the CLI, run:
lamin save sample.fasta --key sample.fasta
To load an artifact via the CLI into a local cache, run:
lamin load --key sample.fasta
Read more about the CLI: docs.lamin.ai/cli.
Trace data, code & agents¶
The lamindb skill ships with the lamindb package at .agents/skills/. Ask your coding agent to copy it to wherever it reads skills from — .claude/skills/ for Claude Code, .agents/skills/ for GitHub Copilot — so that it automatically tracks agent sessions.
To create a dataset in a script or notebook while tracking source code, inputs, outputs, logs, and environment:
import lamindb as ln
# → connected lamindb: account/instance
ln.track() # track code execution
open("sample.fasta", "w").write(">seq1\nACGT\n") # create dataset
ln.Artifact("sample.fasta", key="sample.fasta").save() # save dataset
ln.finish() # mark run as finished
library(laminr)
ln <- import_module("lamindb")
# → connected lamindb: account/instance
ln$track() # track code execution
writeLines(">seq1\nACGT\n", "sample.fasta") # create dataset
ln$Artifact("sample.fasta", key = "sample.fasta")$save() # save dataset
ln$finish() # mark run as finished
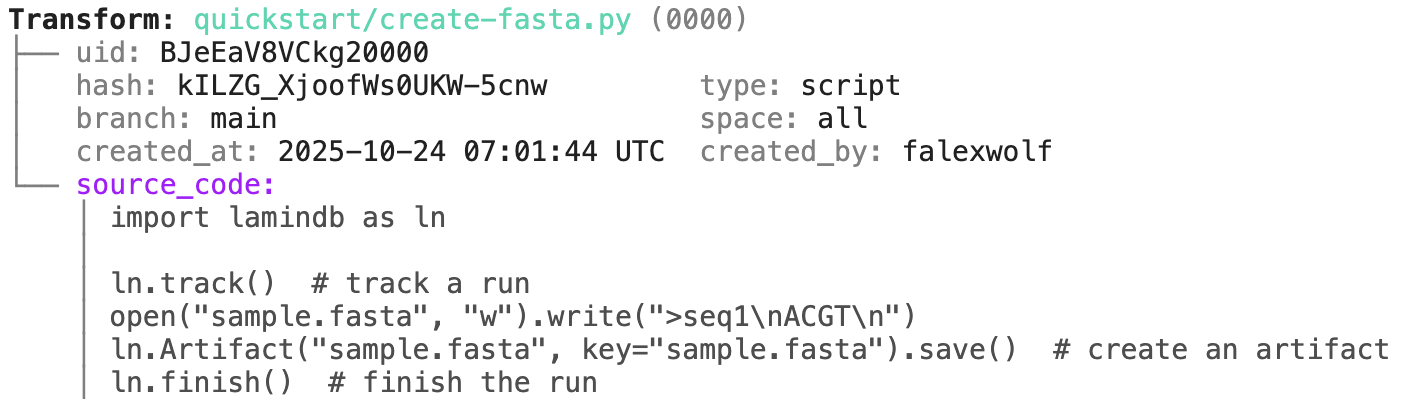
Running this snippet as a script (python create_fasta.py) produces the following data lineage:
artifact = ln.Artifact.get(key="sample.fasta") # get artifact by key
artifact.describe() # context of the artifact
artifact.view_lineage() # fine-grained lineage
artifact <- ln$Artifact$get(key = "sample.fasta") # get artifact by key
artifact$describe() # context of the artifact
artifact$view_lineage() # fine-grained lineage
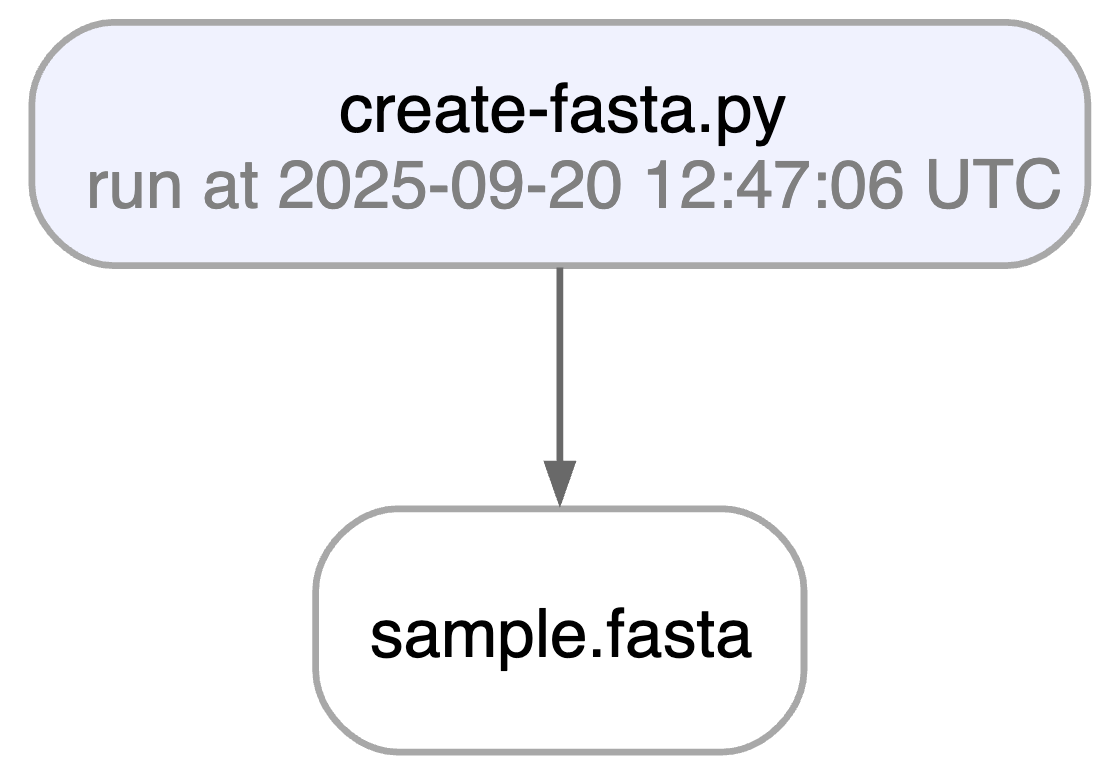
Watch a mini video: youtu.be/yK3ODFZLL1A
Access run & transform.
run = artifact.run # get the run object
transform = artifact.transform # get the transform object
run.describe() # context of the run
run <- artifact$run # get the run object
transform <- artifact$transform # get the transform object
run$describe() # context of the run
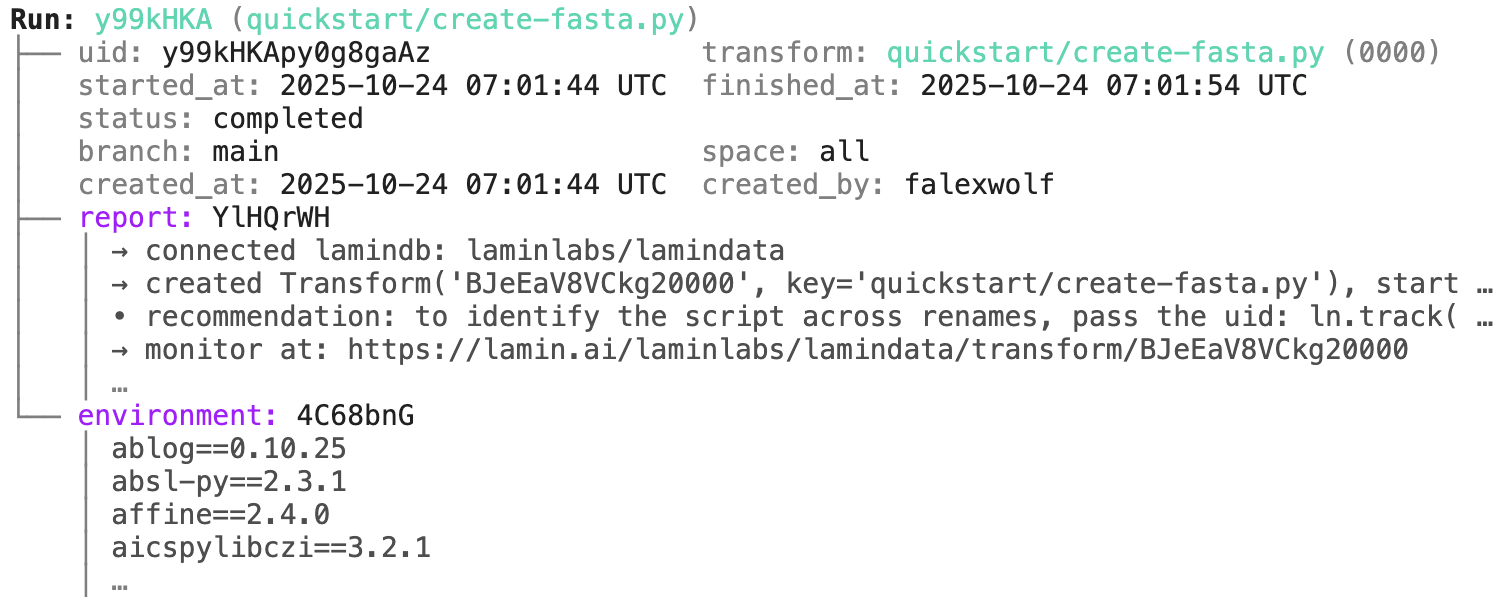
transform.describe() # context of the transform
transform$describe() # context of the transform
Track a project or an agent plan.
Pass a project/artifact to ln.track(), for example:
ln.track(project="My project", plan="./plans/curate-dataset-x.md")
ln$track(project = "My project", plan = "./plans/curate-dataset-x.md")
Note that you have to create a project or save the agent plan in case they don’t yet exist:
# create a project with the CLI
lamin create project "My project"
# save an agent plan with the CLI
lamin save /path/to/.cursor/plans/curate-dataset-x.plan.md
lamin save /path/to/.claude/plans/curate-dataset-x.md
Or in Python:
ln.Project(name="My project").save() # create a project in Python
ln$Project(name = "My project")$save() # create a project in Python
You can track workflows by decorating functions:
import lamindb as ln
@ln.flow()
def create_fasta(fasta_file: str = "sample.fasta"):
open(fasta_file, "w").write(">seq1\nACGT\n") # create dataset
ln.Artifact(fasta_file, key=fasta_file).save() # save dataset
if __name__ == "__main__":
create_fasta()
Beyond what you get for scripts & notebooks, this automatically tracks function & CLI params and integrates well with established Python workflow managers: docs.lamin.ai/track. To integrate advanced bioinformatics pipeline managers like Nextflow, see docs.lamin.ai/pipelines.
A richer example.
Here is an automatically generated re-construction of the project of Schmidt et al. (Science, 2022):
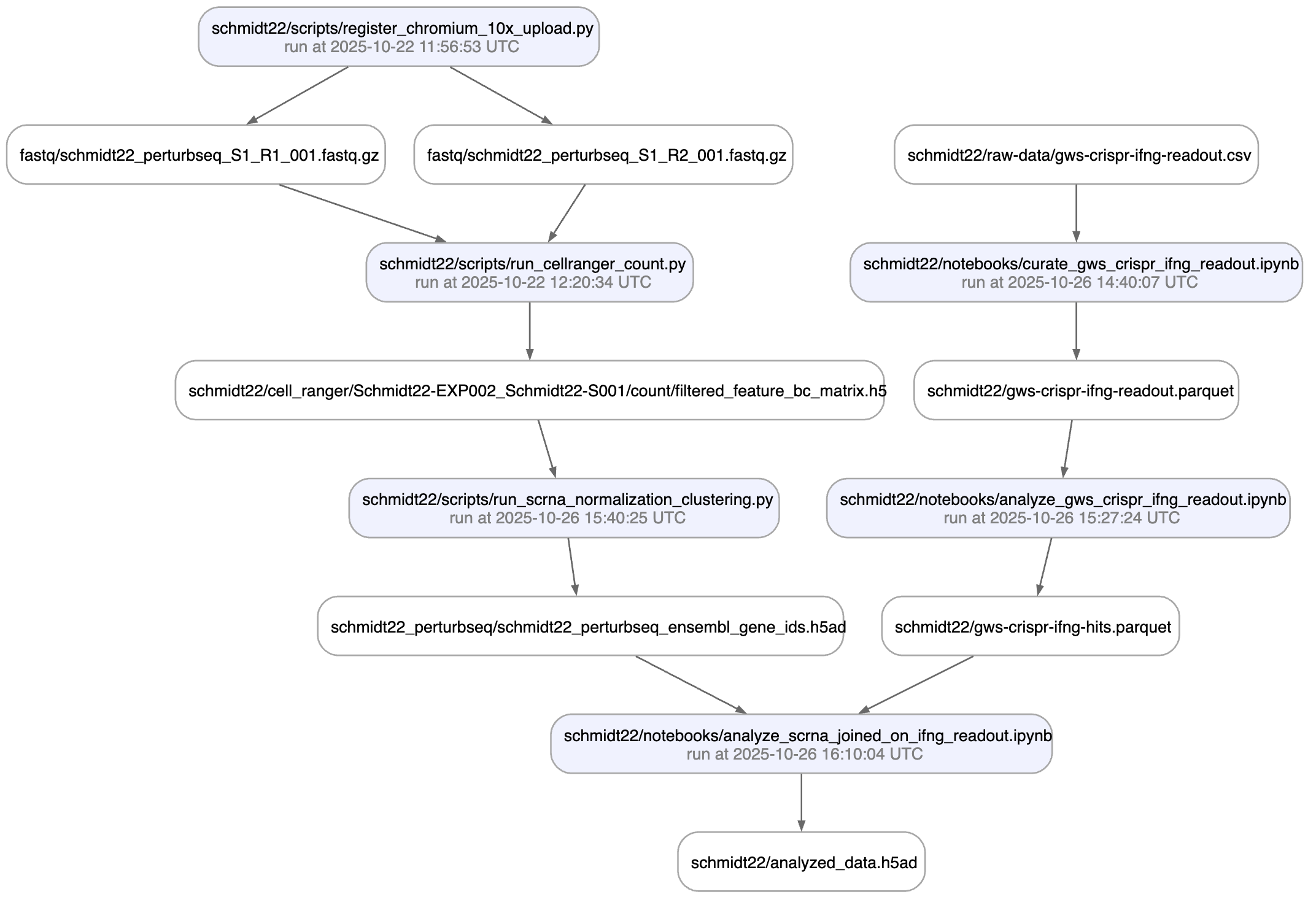
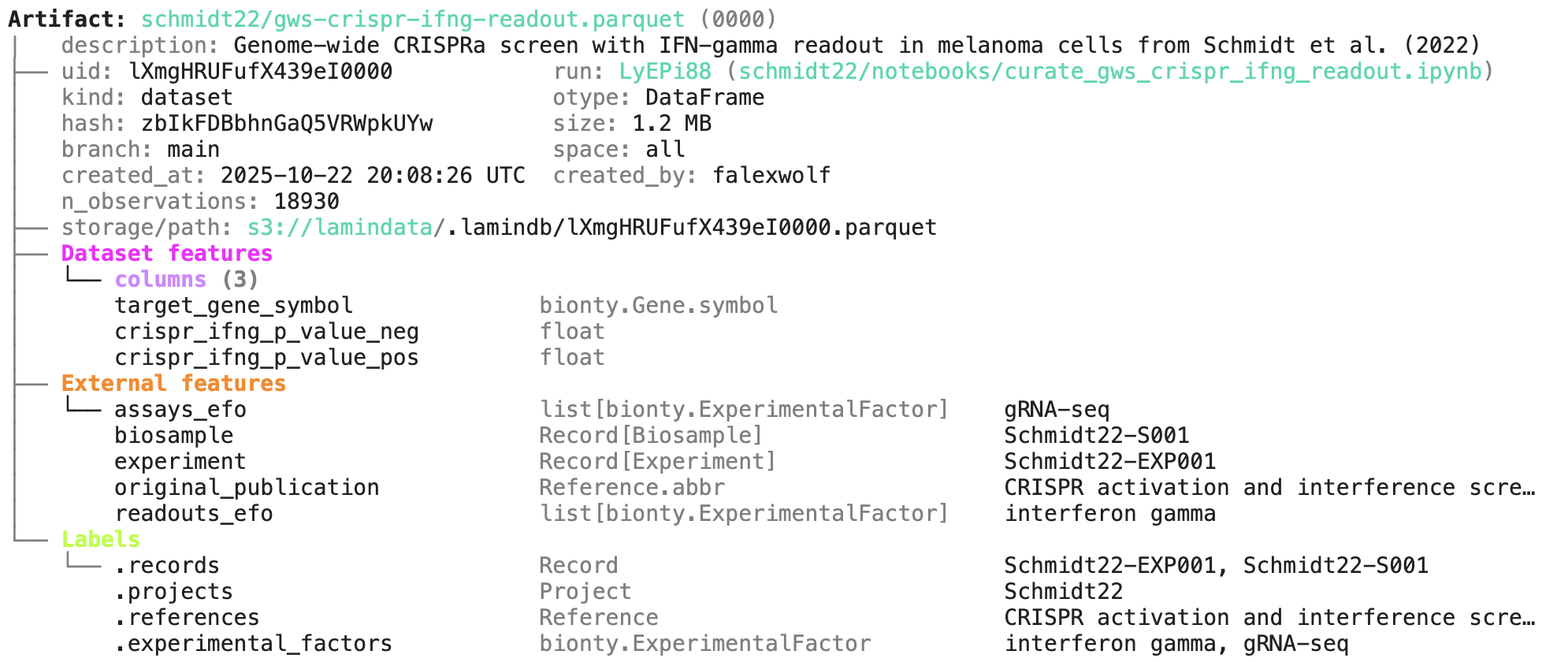
A phenotypic CRISPRa screening result is integrated with scRNA-seq data. Here is the result of the screen input:
Label artifacts¶
You can label an artifact by running:
my_label = ln.ULabel(name="My label").save() # a universal label
project = ln.Project(name="My project").save() # a project label
artifact.ulabels.add(my_label)
artifact.projects.add(project)
my_label <- ln$ULabel(name = "My label")$save() # a universal label
project <- ln$Project(name = "My project")$save() # a project label
artifact$ulabels$add(my_label)
artifact$projects$add(project)
Query for it:
ln.Artifact.filter(ulabels=my_label, projects=project).to_dataframe()
ln$Artifact$filter(ulabels = my_label, projects = project)$to_dataframe()
You can also query by the metadata that lamindb automatically collects:
ln.Artifact.filter(run=run).to_dataframe() # by creating run
ln.Artifact.filter(transform=transform).to_dataframe() # by creating transform
ln.Artifact.filter(size__gt=1e6).to_dataframe() # size greater than 1MB
ln$Artifact$filter(run = run)$to_dataframe() # by creating run
ln$Artifact$filter(transform = transform)$to_dataframe() # by creating transform
ln$Artifact$filter(size__gt = 1e6)$to_dataframe() # size greater than 1MB
If you want to include more information into the resulting dataframe, pass include.
ln.Artifact.to_dataframe(include=["created_by__name", "storage__root"]) # include fields from related registries
ln$Artifact$to_dataframe(include = list("created_by__name", "storage__root")) # include fields from related registries
The query syntax for DB objects and for your default database is the same.
Here is an overview that illustrates how artifacts can be labeled by other entities:

Read more: docs.lamin.ai/organize.
Manage features & records¶
Let’s define some features:
from datetime import date
gc_content = ln.Feature(name="gc_content", dtype=float).save()
experiment_note = ln.Feature(name="experiment_note", dtype=str).save()
experiment_date = ln.Feature(name="experiment_date", dtype=date, coerce=True).save() # accept date strings
datetime <- import_module("datetime")
date <- datetime$date
gc_content <- ln$Feature(name = "gc_content", dtype = "float")$save()
experiment_note <- ln$Feature(name = "experiment_note", dtype = "str")$save()
experiment_date <- ln$Feature(name = "experiment_date", dtype = date, coerce = TRUE)$save() # accept date strings
The most basic thing you can do with features is annotating artifacts, records, or runs with them:
artifact.features.set_values({
gc_content: 0.55,
experiment_note: "Looks great",
experiment_date: "2025-10-24",
})
# query
ln.Artifact.filter(experiment_date == "2025-10-24").to_dataframe(include="features") # query all artifacts annotated with `experiment_date`
artifact$features$set_values(list(
gc_content = 0.55,
experiment_note = "Looks great",
experiment_date = "2025-10-24"
))
# query
ln$Artifact$filter(experiment_date == "2025-10-24")$to_dataframe(include = "features") # query all artifacts annotated with `experiment_date`
You can create records for entities underlying your experiments (samples, perturbations, instruments, etc.):
ln.Record(name="Sample 1", features={gc_content: 0.5}).save()
ln$Record(name = "Sample 1", features = list(gc_content = 0.5))$save()
You can dynamically create registries and relationships of entities via record types:
# create an experiments registry by defining a record type
experiments_registry = ln.Record(name="Experiments", is_type=True).save()
# create a record inside the Experiments registry
ln.Record(name="Experiment 1", type=experiments_registry).save()
# create a feature that links experiments, creating a relationship
experiment = ln.Feature(name="experiment", dtype=experiments_registry).save()
# create a sample record that links the sample to `Experiment 1` via the `experiment` feature
ln.Record(name="Sample 2", features={gc_content: 0.5, experiment: "Experiment 1"}).save()
# export a registry to a dataframe
experiments_registry.to_dataframe()
# create an experiments registry by defining a record type
experiments_registry <- ln$Record(name = "Experiments", is_type = TRUE)$save()
# create a record inside the Experiments registry
ln$Record(name = "Experiment 1", type = experiments_registry)$save()
# create a feature that links experiments, creating a relationship
experiment <- ln$Feature(name = "experiment", dtype = experiments_registry)$save()
# create a sample record that links the sample to `Experiment 1` via the `experiment` feature
ln$Record(name = "Sample 2", features = list(gc_content = 0.5, experiment = "Experiment 1"))$save()
# export a registry to a dataframe
experiments_registry$to_dataframe()
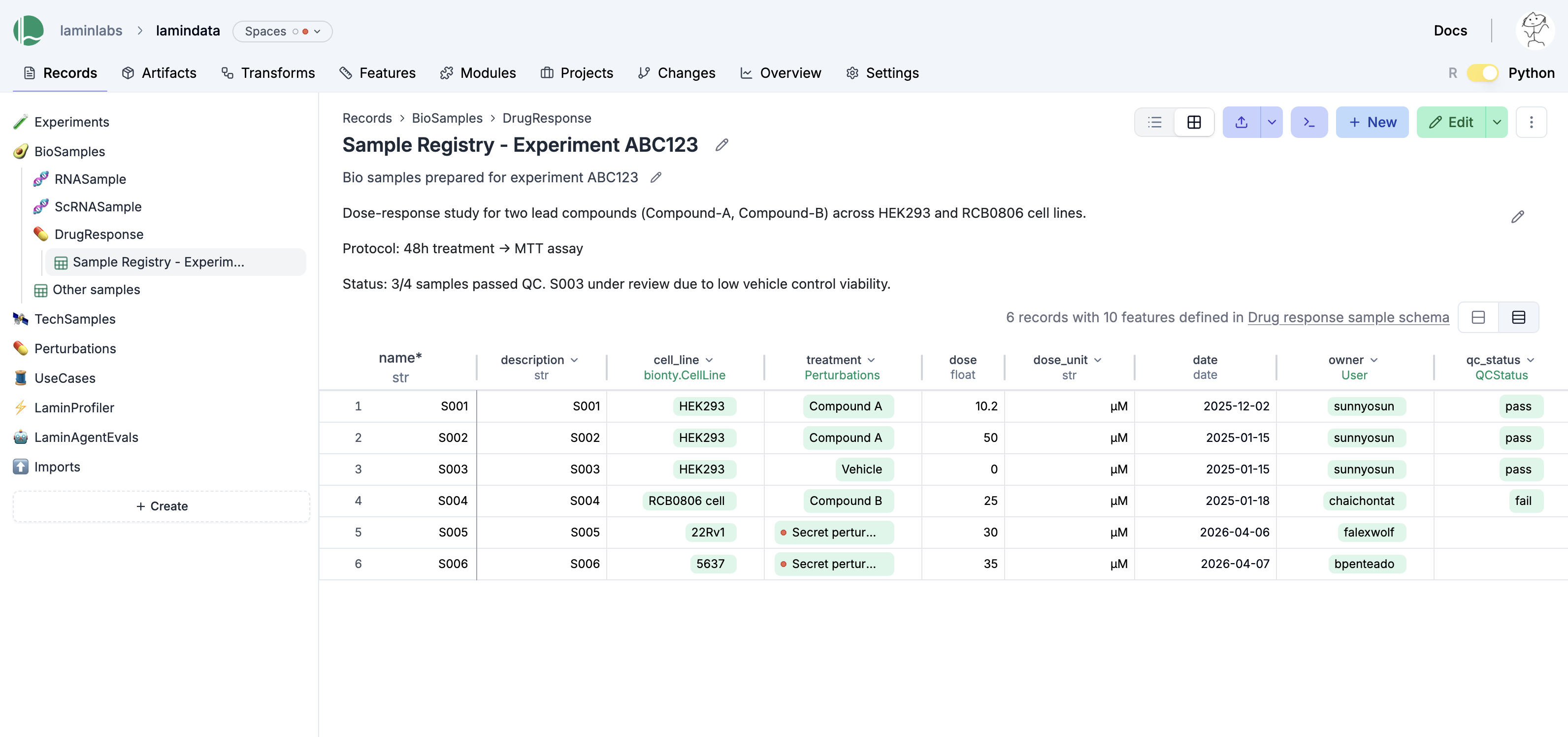
You can edit records like Excel sheets on LaminHub.
Lakehouse¶
Here is how you ingest a DataFrame:
import pandas as pd
df = pd.DataFrame({
"sequence_str": ["ACGT", "TGCA"],
"gc_content": [0.55, 0.54],
"experiment_note": ["Looks great", "Ok"],
"experiment_date": [date(2025, 10, 24), date(2025, 10, 25)],
})
ln.Artifact.from_dataframe(df, key="my_datasets/sequences.parquet").save() # no validation
pd <- import_module("pandas")
df <- pd$DataFrame(list(
sequence_str = list("ACGT", "TGCA"),
gc_content = list(0.55, 0.54),
experiment_note = list("Looks great", "Ok"),
experiment_date = list(date(2025L, 10L, 24L), date(2025L, 10L, 25L))
))
ln$Artifact$from_dataframe(df, key = "my_datasets/sequences.parquet")$save() # no validation
To validate & annotate the content of the dataframe, use the built-in schema valid_features:
ln.Feature(name="sequence_str", dtype=str).save() # define a remaining feature
artifact = ln.Artifact.from_dataframe(
df,
key="my_datasets/sequences.parquet",
schema="valid_features" # validate columns against features
).save()
artifact.describe()
ln$Feature(name = "sequence_str", dtype = "str")$save() # define a remaining feature
artifact <- ln$Artifact$from_dataframe(
df,
key = "my_datasets/sequences.parquet",
schema = "valid_features" # validate columns against features
)$save()
artifact$describe()
Watch a mini video: youtu.be/Ji6E7hTnReQ
You can filter for datasets by schema and then launch distributed queries or batch load distributed datasets. For tables, see: docs.lamin.ai/tables. For arrays, see: docs.lamin.ai/arrays.
To validate an AnnData, call:
import anndata as ad
import numpy as np
import pandas as pd
adata = ad.AnnData(
X=np.ones((21, 10)),
obs=pd.DataFrame({'cell_type_by_model': ['T cell', 'B cell', 'NK cell'] * 7}),
var=pd.DataFrame(index=[f'ENSG{i:011d}' for i in range(10)])
)
artifact = ln.Artifact.from_anndata(
adata,
key="my_datasets/scrna.h5ad",
schema="ensembl_gene_ids_and_valid_features_in_obs"
).save()
artifact.describe()
ad <- import_module("anndata")
np <- import_module("numpy")
pd <- import_module("pandas")
adata <- anndata::AnnData(
X = np$ones((21, 10)),
obs = pd$DataFrame(list(cell_type_by_model = rep(list('T cell', 'B cell', 'NK cell'), 7))),
var = pd$DataFrame(index = sprintf("ENSG%010d", 1:10))
)
artifact <- ln$Artifact$from_anndata(
adata,
key = "my_datasets/scrna.h5ad",
schema = "ensembl_gene_ids_and_valid_features_in_obs"
)$save()
artifact$describe()
To validate a SpatialData or any other array-like dataset, you need to construct a Schema. You can do this by composing simple pandera-style schemas: docs.lamin.ai/curate.
Branching & versioning¶
LaminDB co-versions code and datasets for you.
If edit and run the create_fasta.py script, you’ll automatically create a new version of the transform and the sample.fasta artifact.
The edited script
# create_fasta.py
import lamindb as ln
ln.track()
open("sample.fasta", "w").write(">seq1\nTGCA\n") # a new sequence
ln.Artifact("sample.fasta", key="sample.fasta", features={"experiment": "Experiment 1"}).save() # annotate with the new experiment
ln.finish()
# create_fasta$py
library(laminr)
ln <- import_module("lamindb")
ln$track()
writeLines(">seq1\nTGCA\n", "sample.fasta") # a new sequence
ln$Artifact("sample.fasta", key = "sample.fasta", features = list(experiment = "Experiment 1"))$save() # annotate with the new experiment
ln$finish()
artifact_latest = ln.Artifact.get(key="sample.fasta") # pass version for a previous version: ln.Artifact.get(key="sample.fasta", version="1.0")
artifact_latest.versions.to_dataframe() # all versions of that artifact
artifact_latest.transform.versions.to_dataframe() # all versions of the transform that created the artifact
artifact_latest <- ln$Artifact$get(key = "sample.fasta") # pass version for a previous version: ln$Artifact$get(key = "sample.fasta", version = "1.0")
artifact_latest$versions$to_dataframe() # all versions of that artifact
artifact_latest$transform$versions$to_dataframe() # all versions of the transform that created the artifact
To isolate changes, create a contribution branch and switch to it as in git:
lamin switch -c my_branch
To merge a contribution branch into main, run:
lamin switch main # switch to the main branch
lamin merge my_branch # merge contribution branch into main
Read more: docs.lamin.ai/manage-changes.
Watch a mini video: youtu.be/rzRwcMj6-fc
Data sharing¶
To share data in a lineage-aware way, transfer objects from a source database to your default database:
db = ln.DB("laminlabs/lamindata")
artifact = db.Artifact.get(key="example_datasets/mini_immuno/dataset1.h5ad")
artifact.save()
db <- ln$DB("laminlabs/lamindata")
artifact <- db$Artifact$get(key = "example_datasets/mini_immuno/dataset1.h5ad")
artifact$save()
This is zero-copy for the artifact’s data in storage. Read more: docs.lamin.ai/transfer.
Ontologies¶
Plugin bionty gives you >20 public ontologies as SQLRecord registries. This was used to validate the ENSG ids in the adata just before.
import bionty as bt
bt.CellType.import_source() # import the default ontology
bt.CellType.to_dataframe() # your extensible cell type ontology in a simple registry
bt <- import_module("bionty")
bt$CellType$import_source() # import the default ontology
bt$CellType$to_dataframe() # your extensible cell type ontology in a simple registry
You can then create objects, e.g. for labeling, analogous to ULabel, Project, or Record:
t_cell = bt.CellType.get(name="T cell")
artifact.cell_types.add(t_cell)
t_cell <- bt$CellType$get(name = "T cell")
artifact$cell_types$add(t_cell)
Read more: docs.lamin.ai/manage-ontologies.
Watch a mini video: youtu.be/3vpWjHj3Kw8
Manage notes¶
When in your development directory, you can save markdown files as records:
lamin save <topic>/<my-note.md>