Nextflow 
 ¶
¶
There are two ways of tracking Nextflow pipeline runs with their inputs and outputs in LaminDB.
nf-lamin¶
The nf-lamin Nextflow plugin works without modifying pipeline code via LaminHub’s REST API.
Option A: environment variables (no config file needed):
export LAMIN_CURRENT_INSTANCE="your-org/your-instance"
export LAMIN_API_KEY="<your-lamin-api-key>"
nextflow run -plugins nf-lamin <your-pipeline>
Option B: Nextflow secrets + config file:
Store your API key as a Nextflow secret:
nextflow secrets set LAMIN_API_KEY <your-lamin-api-key>
Create a lamin.config:
%%groovy
plugins {
id 'nf-lamin'
}
lamin {
instance = "your-org/your-instance"
api_key = secrets.LAMIN_API_KEY
}
Then run your pipeline with the config:
nextflow run <your-pipeline> -c lamin.config
After the run, explore the tracked data in LaminHub or via the Python SDK:
import lamindb as ln
ln.Run.get("your-run-uid")
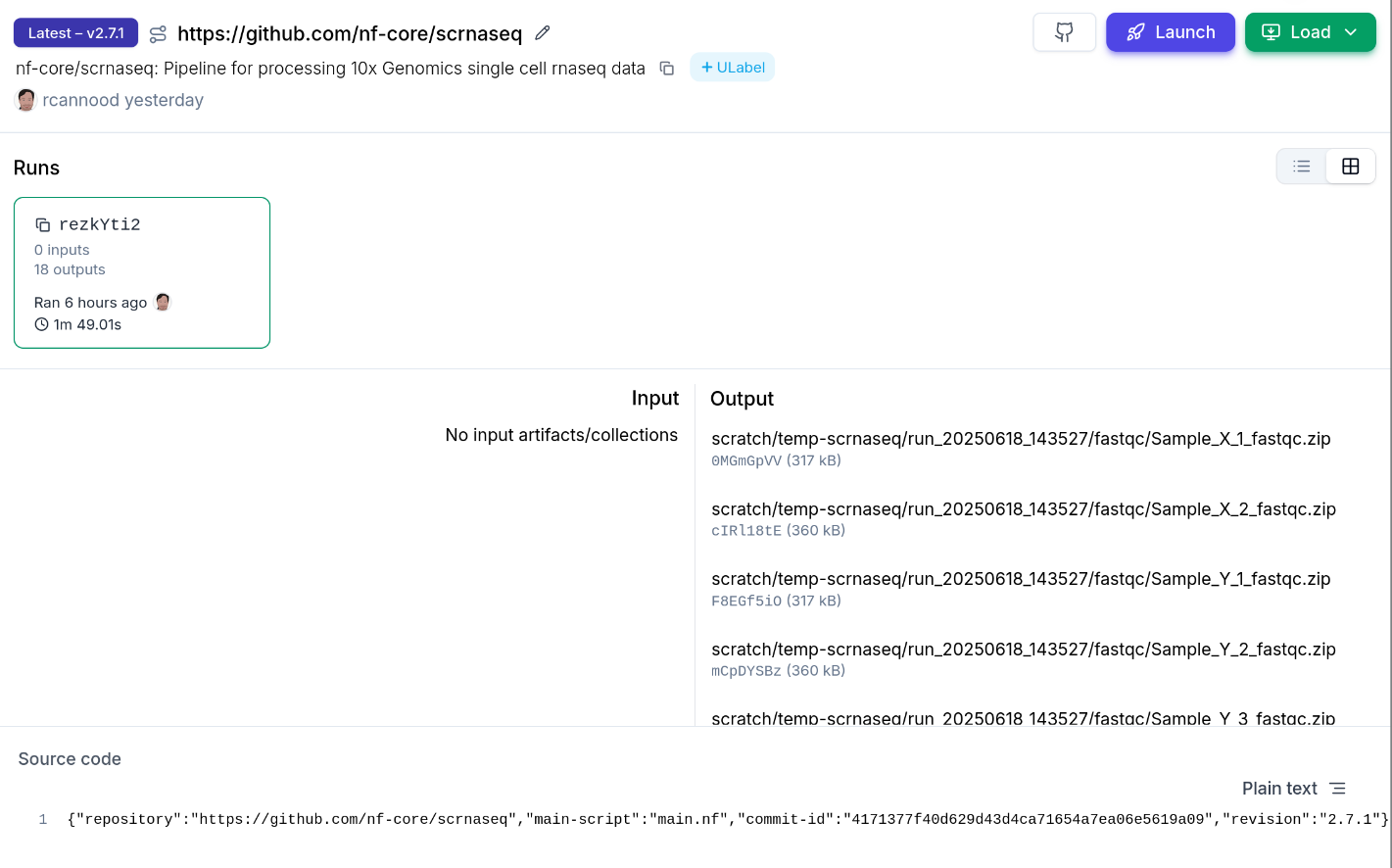
Runs executed with -with-tower or launched from Seqera Platform additionally get run.reference set
to the Platform watch URL, with run.reference_type set to "Seqera".
→ See Nextflow: nf-laminfor the fullnf-lamin` configuration reference.
→ See Examples for ready-to-run examples for existing pipelines.
Post-run scripts¶
You can register runs manually without using the nf-lamin plugin using LaminDB in a Python post-run script. First run the pipeline:
# the test profile uses all downloaded input files as an input
!nextflow run nf-core/scrnaseq -r 4.0.0 -profile docker,test -resume --outdir scrnaseq_output
Show code cell output
N E X T F L O W ~ version 26.04.6
Pulling nf-core/scrnaseq:4.0.0 ...
WARN: Cannot read project manifest -- Cause: Config parsing failed
downloaded from https://github.com/nf-core/scrnaseq.git
Error nextflow.config:232:28: Invalid include source: '/home/runner/.nextflow/assets/.repos/nf-core/scrnaseq/clones/e0ddddbff9d8b8c2421c67ff07449a06f9ca02d2/conf/test_multiome.config'
│ 232 | test_multiome { includeConfig 'conf/test_multiome.config'
╰ | ^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^
Error nextflow.config:373:32: `manifest` is not defined
│ 373 | \033[0;35m nf-core/scrnaseq ${manifest.version}\033[0m
╰ | ^^^^^^^^
Error nextflow.config:376:26: `manifest` is not defined
│ 376 | afterText = """${manifest.doi ? "\n* The pipeline\n" : ""}${ma
╰ | ^^^^^^^^
Error nextflow.config:376:69: `manifest` is not defined
│ 376 | ? "\n* The pipeline\n" : ""}${manifest.doi.tokenize(",").collect { "
╰ | ^^^^^^^^
Error nextflow.config:376:186: `manifest` is not defined
│ 376 | doi.org/','')}"}.join("\n")}${manifest.doi ? "\n" : ""}
╰ | ^^^^^^^^
Error nextflow.config:385:22: `validation` is not defined
│ 385 | beforeText = validation.help.beforeText
╰ | ^^^^^^^^^^
Error nextflow.config:386:21: `validation` is not defined
│ 386 | afterText = validation.help.afterText
╰ | ^^^^^^^^^^
ERROR ~ Config parsing failed
-- Check '.nextflow.log' file for details
Example: nf-core/scrnaseq
After the run is complete, use a post-run script to register inputs and outputs in LaminDB:
import argparse
import lamindb as ln
import json
import re
from pathlib import Path
from lamin_utils import logger
def parse_arguments() -> argparse.Namespace:
parser = argparse.ArgumentParser()
parser.add_argument("--input", type=str, required=True)
parser.add_argument("--output", type=str, required=True)
return parser.parse_args()
def register_pipeline_io(input_dir: str, output_dir: str, run: ln.Run) -> None:
"""Register input and output artifacts for an `nf-core/scrnaseq` run."""
input_artifacts = ln.Artifact.from_dir(input_dir, run=False)
ln.save(input_artifacts)
run.input_artifacts.set(input_artifacts)
ln.Artifact(f"{output_dir}/multiqc", description="multiqc report", run=run).save()
ln.Artifact(
f"{output_dir}/star/mtx_conversions/combined_filtered_matrix.h5ad",
key="filtered_count_matrix.h5ad",
run=run,
).save()
def register_pipeline_metadata(output_dir: str, run: ln.Run) -> None:
"""Register nf-core run metadata stored in the 'pipeline_info' folder."""
ulabel = ln.ULabel(name="nextflow").save()
run.transform.ulabels.add(ulabel)
# nextflow run id
content = next(Path(f"{output_dir}/pipeline_info").glob("execution_report_*.html")).read_text()
match = re.search(r"run id \[([^\]]+)\]", content)
nextflow_id = match.group(1) if match else ""
run.reference = nextflow_id
run.reference_type = "nextflow_id"
# completed at
completion_match = re.search(r'<span id="workflow_complete">([^<]+)</span>', content)
if completion_match:
from datetime import datetime
timestamp_str = completion_match.group(1).strip()
run.finished_at = datetime.strptime(timestamp_str, "%d-%b-%Y %H:%M:%S")
# execution report and software versions
for file_pattern, description, run_attr in [
("execution_report*", "execution report", "report"),
("nf_core_*_software*", "software versions", "environment"),
]:
matching_files = list(Path(f"{output_dir}/pipeline_info").glob(file_pattern))
if not matching_files:
logger.warning(f"No files matching '{file_pattern}' in pipeline_info")
continue
artifact = ln.Artifact(
matching_files[0],
description=f"nextflow run {description} of {nextflow_id}",
visibility=0,
run=False,
).save()
setattr(run, run_attr, artifact)
# nextflow run parameters
params_path = next(Path(f"{output_dir}/pipeline_info").glob("params*"))
with params_path.open() as params_file:
params = json.load(params_file)
ln.Param(name="params", dtype="dict").save()
run.features.add_values({"params": params})
run.save()
args = parse_arguments()
scrnaseq_transform = ln.Transform(
key="scrna-seq",
version="4.0.0",
type="pipeline",
reference="https://github.com/nf-core/scrnaseq",
).save()
run = ln.Run(transform=scrnaseq_transform).save()
register_pipeline_io(args.input, args.output, run)
register_pipeline_metadata(args.output, run)
!python nextflow/register_scrnaseq_run.py --input scrnaseq_input --output scrnaseq_output
Show code cell output
/home/runner/work/nf-lamin/nf-lamin/docs/nextflow/register_scrnaseq_run.py:77: DeprecationWarning: `type` argument of transform was renamed to `kind` and will be removed in a future release.
scrnaseq_transform = ln.Transform(
Traceback (most recent call last):
File "/home/runner/work/nf-lamin/nf-lamin/docs/nextflow/register_scrnaseq_run.py", line 77, in <module>
scrnaseq_transform = ln.Transform(
~~~~~~~~~~~~^
key="scrna-seq",
^^^^^^^^^^^^^^^^
...<2 lines>...
reference="https://github.com/nf-core/scrnaseq",
^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^
).save()
^
File "/opt/hostedtoolcache/Python/3.14.6/x64/lib/python3.14/site-packages/lamindb/models/transform.py", line 346, in __init__
candidates_for_revises.filter(branch_id=target_branch_id).first()
~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~^^
File "/opt/hostedtoolcache/Python/3.14.6/x64/lib/python3.14/site-packages/lamindb/models/query_set.py", line 1401, in first
if len(self) == 0:
~~~^^^^^^
File "/opt/hostedtoolcache/Python/3.14.6/x64/lib/python3.14/site-packages/django/db/models/query.py", line 368, in __len__
self._fetch_all()
~~~~~~~~~~~~~~~^^
File "/opt/hostedtoolcache/Python/3.14.6/x64/lib/python3.14/site-packages/django/db/models/query.py", line 1954, in _fetch_all
self._result_cache = list(self._iterable_class(self))
~~~~^^^^^^^^^^^^^^^^^^^^^^^^^^^^
File "/opt/hostedtoolcache/Python/3.14.6/x64/lib/python3.14/site-packages/django/db/models/query.py", line 93, in __iter__
results = compiler.execute_sql(
chunked_fetch=self.chunked_fetch, chunk_size=self.chunk_size
)
File "/opt/hostedtoolcache/Python/3.14.6/x64/lib/python3.14/site-packages/django/db/models/sql/compiler.py", line 1623, in execute_sql
cursor.execute(sql, params)
~~~~~~~~~~~~~~^^^^^^^^^^^^^
File "/opt/hostedtoolcache/Python/3.14.6/x64/lib/python3.14/site-packages/django/db/backends/utils.py", line 79, in execute
return self._execute_with_wrappers(
~~~~~~~~~~~~~~~~~~~~~~~~~~~^
sql, params, many=False, executor=self._execute
^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^
)
^
File "/opt/hostedtoolcache/Python/3.14.6/x64/lib/python3.14/site-packages/django/db/backends/utils.py", line 92, in _execute_with_wrappers
return executor(sql, params, many, context)
File "/opt/hostedtoolcache/Python/3.14.6/x64/lib/python3.14/site-packages/lamindb_setup/core/django.py", line 56, in error_no_instance_wrapper
raise CurrentInstanceNotConfigured
lamindb_setup.errors.CurrentInstanceNotConfigured: No instance is connected! Call
- CLI: lamin connect / lamin init
- Python: ln.connect() / ln.setup.init()
- R: ln$connect() / ln$setup$init()
Such a script can also be triggered from a serverless environment (e.g., AWS Lambda).