Nextflow  ¶
¶
There are several ways to track Nextflow pipeline runs and artifacts in LaminDB.
Using nf-lamin (recommended)¶
The nf-lamin Nextflow plugin automatically tracks transforms, runs, and artifacts without modifying pipeline code. It requires a LaminHub account.
1. Store your Lamin API key as a Nextflow secret:
nextflow secrets set LAMIN_API_KEY <your-lamin-api-key>
2. Add the plugin to your nextflow.config:
plugins {
id 'nf-lamin'
}
lamin {
instance = "your-org/your-instance"
api_key = secrets.LAMIN_API_KEY
}
3. Run your pipeline:
nextflow run <your-pipeline>
After the run, explore the tracked data in LaminHub or via the Python SDK:
import lamindb as ln
ln.Run.get("your-run-uid")
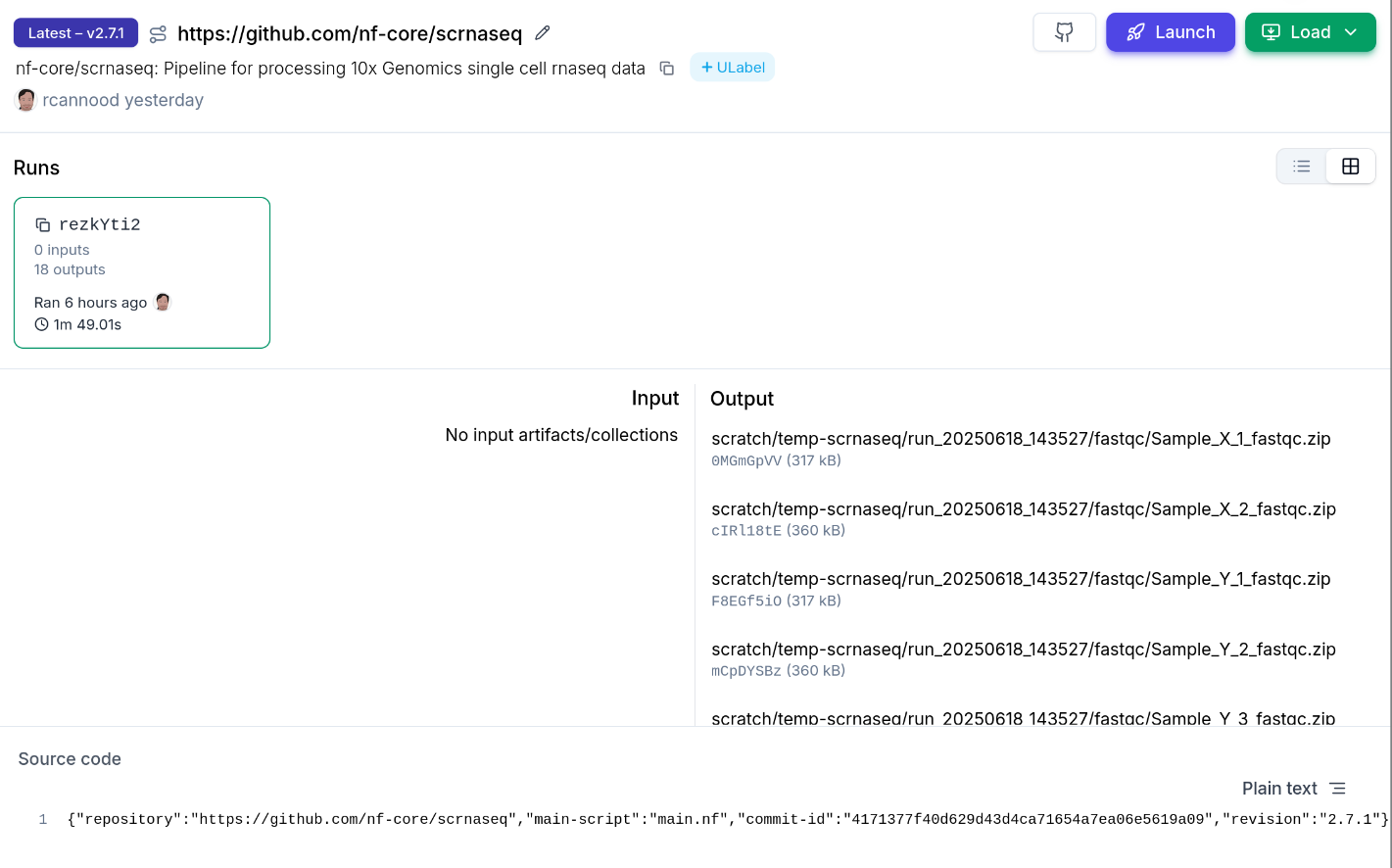
→ See Nextflow: nf-laminfor the fullnf-lamin` configuration reference.
→ See Examples for ready-to-run examples for existing pipelines.
Using a post-run script¶
If you want to use Nextflow with LaminDB but without the nf-lamin plugin, you can register runs manually with a Python post-run script.
Example: nf-core/scrnaseq post-run registration
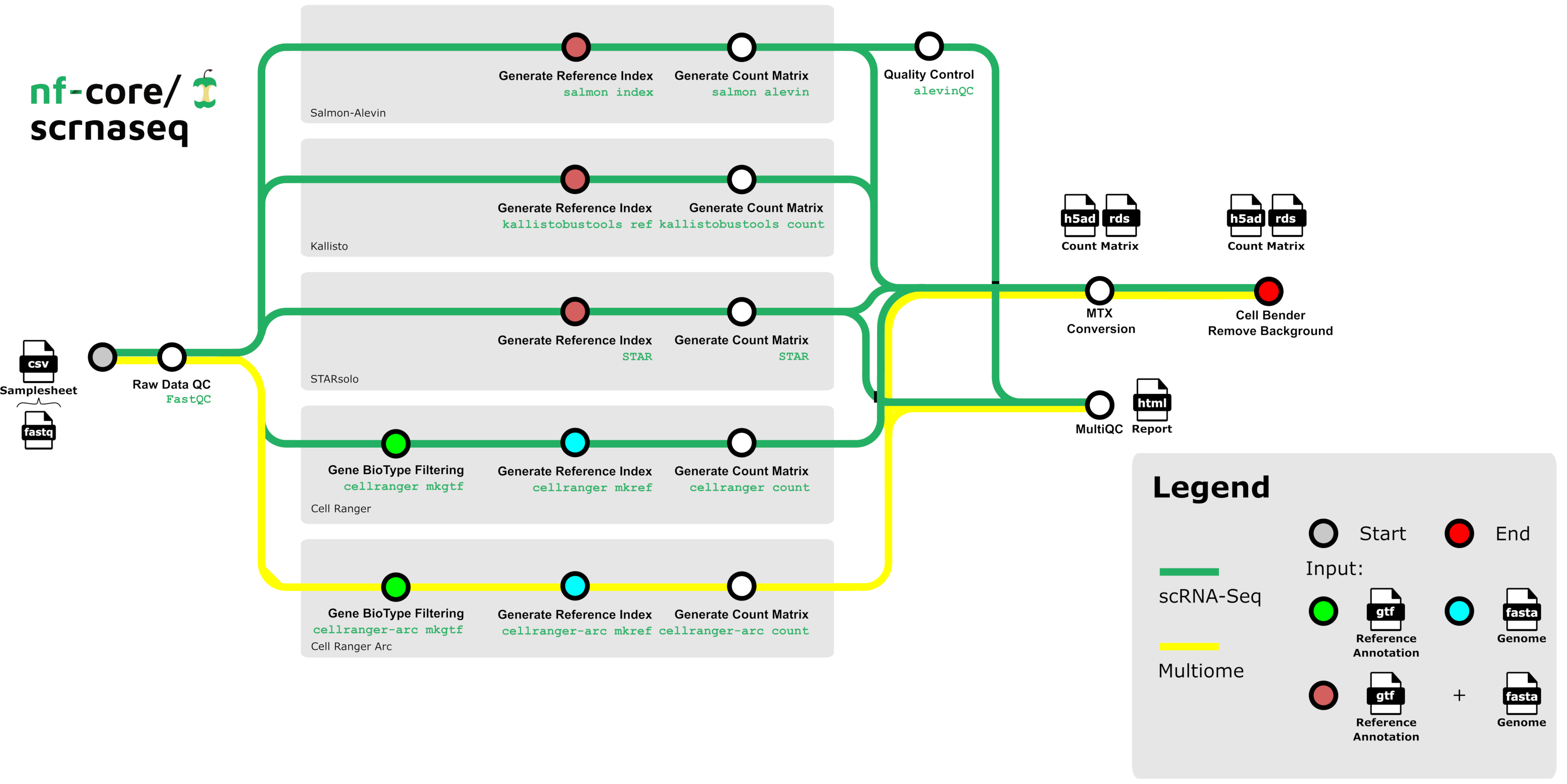
After running the pipeline, a Python script registers inputs & outputs in LaminDB:
import argparse
import lamindb as ln
import json
import re
from pathlib import Path
from lamin_utils import logger
def parse_arguments() -> argparse.Namespace:
parser = argparse.ArgumentParser()
parser.add_argument("--input", type=str, required=True)
parser.add_argument("--output", type=str, required=True)
return parser.parse_args()
def register_pipeline_io(input_dir: str, output_dir: str, run: ln.Run) -> None:
"""Register input and output artifacts for an `nf-core/scrnaseq` run."""
input_artifacts = ln.Artifact.from_dir(input_dir, run=False)
ln.save(input_artifacts)
run.input_artifacts.set(input_artifacts)
ln.Artifact(f"{output_dir}/multiqc", description="multiqc report", run=run).save()
ln.Artifact(
f"{output_dir}/star/mtx_conversions/combined_filtered_matrix.h5ad",
key="filtered_count_matrix.h5ad",
run=run,
).save()
def register_pipeline_metadata(output_dir: str, run: ln.Run) -> None:
"""Register nf-core run metadata stored in the 'pipeline_info' folder."""
ulabel = ln.ULabel(name="nextflow").save()
run.transform.ulabels.add(ulabel)
# nextflow run id
content = next(Path(f"{output_dir}/pipeline_info").glob("execution_report_*.html")).read_text()
match = re.search(r"run id \[([^\]]+)\]", content)
nextflow_id = match.group(1) if match else ""
run.reference = nextflow_id
run.reference_type = "nextflow_id"
# completed at
completion_match = re.search(r'<span id="workflow_complete">([^<]+)</span>', content)
if completion_match:
from datetime import datetime
timestamp_str = completion_match.group(1).strip()
run.finished_at = datetime.strptime(timestamp_str, "%d-%b-%Y %H:%M:%S")
# execution report and software versions
for file_pattern, description, run_attr in [
("execution_report*", "execution report", "report"),
("nf_core_*_software*", "software versions", "environment"),
]:
matching_files = list(Path(f"{output_dir}/pipeline_info").glob(file_pattern))
if not matching_files:
logger.warning(f"No files matching '{file_pattern}' in pipeline_info")
continue
artifact = ln.Artifact(
matching_files[0],
description=f"nextflow run {description} of {nextflow_id}",
visibility=0,
run=False,
).save()
setattr(run, run_attr, artifact)
# nextflow run parameters
params_path = next(Path(f"{output_dir}/pipeline_info").glob("params*"))
with params_path.open() as params_file:
params = json.load(params_file)
ln.Param(name="params", dtype="dict").save()
run.features.add_values({"params": params})
run.save()
args = parse_arguments()
scrnaseq_transform = ln.Transform(
key="scrna-seq",
version="4.0.0",
type="pipeline",
reference="https://github.com/nf-core/scrnaseq",
).save()
run = ln.Run(transform=scrnaseq_transform).save()
register_pipeline_io(args.input, args.output, run)
register_pipeline_metadata(args.output, run)
Run it with:
python register_scrnaseq_run.py --input scrnaseq_input --output scrnaseq_output
If need be, such a script can be deployed via a serverless environment trigger (e.g., AWS Lambda).