lamindb.Feature  ¶
¶
- class lamindb.Feature(name: str, dtype: SimpleDtype | SimpleDtypeStr | ULabel | Record | Registry | list[Registry] | FieldAttr, type: Feature | None = None, is_type: bool = False, unit: str | None = None, description: str | None = None, synonyms: str | None = None, nullable: bool | None = None, default_value: Any | None = None, coerce: bool | None = None, cat_filters: dict[str, SQLRecord | bool | str] | None = None, branch: Branch | None = None, space: Space | None = None)¶
- class lamindb.Feature(*, name: str, is_type: Literal[True], description: str | None = None)
Bases:
SQLRecord,HasType,CanCurate,HasSynonyms,TracksRun,TracksUpdatesMeasurable properties such as columns of a sheet.
Features index variables across datasets to enable querying by dimensions (Query & search ).

- Parameters:
name –
strName of the feature, typically a column name.dtype –
SimpleDtype | ULabel | Record | Registry | list[Registry] | FieldAttrTypes orULabelorRecordobjects representing types. SeeSimpleDtypeStr.type –
Feature | None = NoneA feature type, seetype.is_type –
bool = FalseWhether this feature is a type, seeis_type.unit –
str | None = NoneUnit of measure, ideally SI ("m","s","kg", etc.) or"normalized"etc.description –
str | None = NoneA description.synonyms –
str | None = NoneBar-separated synonyms.nullable –
bool = TrueWhether the feature can have null-like values (None,pd.NA,NaN, etc.), seenullable.default_value –
Any | None = NoneDefault value for the feature.coerce –
bool | None = NoneWhenTrue, attempts to coerce values to the specified dtype during validation, seecoerce. Defaults toFalseunlessis_typeisTrue.cat_filters –
dict[str, SQLRecord | bool | str] | None = NoneSubset a registry by additional filters to define valid categories.branch –
Branch | None = NoneA branch. IfNone, uses the current branch.space –
Space | None = NoneA space. IfNone, uses the current space.
See also
Examples¶
Features with simple data types:
ln.Feature(name="sample_note", dtype=str).save() ln.Feature(name="temperature_in_celsius", dtype=float).save() ln.Feature(name="read_count", dtype=int).save()
A categorical feature measuring labels managed in the
ULabelregistry:ln.Feature(name="sample", dtype=ln.ULabel).save()
Restrict a categorical feature to a specific
ULabeltype, here a perturbations registry:perturbation_registry = ln.ULabel(name="Perturbations", is_type=True).save() ln.Feature(name="perturbation", dtype=perturbation_registry).save()
Restrict a categorical feature to a
Recordtype, here an experiments registry:experiments_registry = ln.Record(name="Experiments", is_type=True).save() ln.Feature(name="experiment", dtype=experiments_registry).save()
Restrict a categorical feature to the
bt.CellTyperegistry:ln.Feature(name="cell_type_by_expert", dtype=bt.CellType).save() # expert annotation ln.Feature(name="cell_type_by_model", dtype=bt.CellType).save() # model annotation
Categoricals define relationships.
For example, when passing
ULabeltodtypeinFeature(), one relates the new feature to theULabelregistry.Scope a feature with a feature type to distinguish the same feature name across different contexts:
abc_feature_type = ln.Feature(name="ABC", is_type=True).save() # ABC could reference a schema, a project, a team, etc. ln.Feature(name="concentration_nM", dtype=float, type=abc_feature_type).save() xyz_feature_type = ln.Feature(name="XYZ", is_type=True).save() # XYZ could reference a schema, a project, a team, etc. ln.Feature(name="concentration_nM", dtype=float, type=xyz_feature_type).save() # calling .save() again with the same name and type returns the existing feature ln.Feature(name="concentration_nM", dtype=float, type=xyz_feature_type).save()
Annotate an artifact with features (works identically for records and runs):
artifact.features.set_values({ "temperature_in_celsius": 37.5, "sample_note": "Control sample", })
Query artifacts/records/runs by features:
ln.Artifact.filter(features__name="temperature_in_celsius") # artifacts with this feature ln.Artifact.filter(temperature_in_celsius__gt=37) # artifacts where temperature > 37
Disambiguate duplicate feature names by querying with a
Featureobject:feature = ln.Feature.get(name="my_ambig_name", type__name="my_feature_type") ln.Artifact.filter(feature == "hello") # instead of my_ambig_name="hello"
A list
dtype:ln.Feature( name="cell_types", dtype=list[bt.CellType], # or list[str] for a list of strings ).save()
A path
dtype:ln.Feature( name="image_path", dtype="path", # will be validated as `str` ).save()
Restrict categories via filters:
# restrict diseases to those matching a specific ontology version source = bt.Source.get(name="My ontology") # a registry for ontology versions ln.Feature( name="disease", dtype=bt.Disease, cat_filters={"source": source}, ).save() # restrict artifacts to those matching a specific schema schema = ln.Schema.get(name="my-schema") ln.Feature( name="valid_artifact", dtype=ln.Artifact, cat_filters={"schema": schema}, ).save() # restrict records to sheets with a shared schema and type sample_type = ln.Record.get(name="Samples") schema = ln.Schema.get(name="my_sample_schema") ln.Feature( name="samplesheet", dtype=sample_type, cat_filters={"is_type": True, "schema": schema}, ).save()
A feature accepting multiple categorical types - a union type:
ln.Feature( name="cell_types", dtype=[bt.Tissue.ontology_id, bt.CellType.ontology_id] ).save()
Notes¶
Features can define validation constraints for individual dataset dimensions. Here is an example where two flow cytometry datasets measure cell markers like
CD4andCD8Aand metadata likesampleandcell_type:
For more, read Curate datasets or Query arrays in storage .
Features work across artifacts, records, and runs.
Here is how records indexed by the features of a sheet look like on the hub UI:
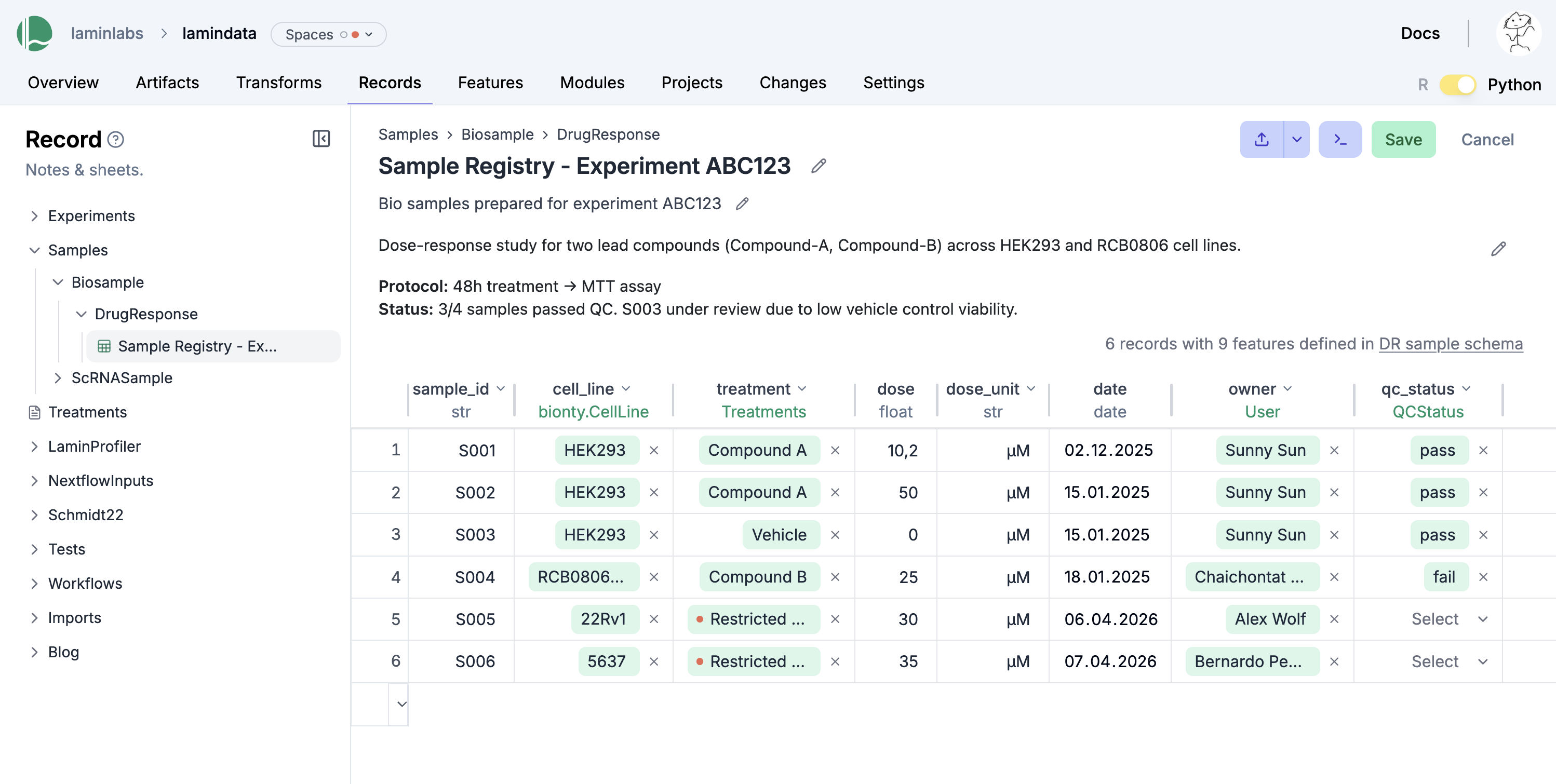
What if my dataset has 40k or more dimensions as in a gene expression dataset?
You don’t bother defining an individual feature for each dimension but instead define a common
dtypefor a set of features along with a constraint for the feature identifier type.For example:
ln.Schema(itype=ln.Feature, dtype=float).save() # use Feature.name as feature identifier type ln.Schema(itype=bt.Gene.ensembl_gene_id, dtype=int).save() # use Gene.ensembl_gene_id as feature identifier type ln.Schema(itype=bt.Protein.uniprot_id, dtype=float).save() # use Protein.uniprot_id as feature identifier type ln.Schema(itype=bt.CellMarker, dtype=float).save() # use CellMarker.name as feature identifier type
In these examples,
biontyregistries are used to leverage biological entities as feature identifiers. If you pass a dataset for validation with this schema, feature identifiers will be validated accordingly.What is the difference between features and labels?
A feature qualifies what is measured, i.e., a numerical or categorical random variable
A label is a measured value of a categorical variable, i.e., a category
Example: When annotating a dataset that measures expression of 30k genes, the gene identifiers serve as feature identifiers, and the features are expression measurements for these genes. When annotating a dataset whose experiment knocked out 3 specific genes, those genes serve as labels of the dataset.
Re-shaping data can introduce ambiguity among features & labels. If this happened, ask yourself what the joint measurement was: a feature qualifies variables in a joint measurement. The canonical data matrix lists jointly measured variables in the columns.
Data types (dtypes)¶
Simple data types. In the table below, the first column shows the object that can be passed to the
dtypeargument ofFeature()orSchema()and the second the string serialization that’s used in the database.dtype
string serialization
pandas
int"int"int64 | int32 | int16 | int8 | uint | ...float"float"float64 | float32 | float16 | float8 | ...str"str"objectbool"bool"boolean | booldatetime"datetime"datetime"datetime64[ns, UTC]""datetime64[ns, UTC]"datetime64[ns, UTC]date"date"object(pandera requires an ISO-format string, convert withdf["date"] = df["date"].dt.date)dict"dict"object"num""num"int | float(“num” is a convenience type forint | float)"path""path"str(pandas does not have a dedicated path type, validated asstr)"url""url"str(pandas does not have a dedicated url type, validated asstr)Categorical and relational data types. For any categorical, you can restrict permissible values to the values defined in a registry. This establishes a relationship.
dtype
string serialization
ln.ULabel"cat[ULabel]"bt.CellType"cat[bionty.CellType]"bt.Disease"cat[bionty.Disease]"ln.Artifact"cat[Artifact]"You can restrict permissible values to instances of
ULabelorRecordtypes, i.e., to dynamic registries.dtype
string serialization
ulabel_type(aULabelwithis_type=True)"cat[ULabel[<uid_of_ulabel_type>]]"record_type(aRecordwithis_type=True)"cat[Record[<uid_of_record_type>]]"You can restrict permissible values by filtering the categorical on fields of its registry.
dtype
cat_filters
string serialization
bt.Disease{"source": source}"cat[bionty.Disease]"ln.Artifact{"schema": schema}"cat[Artifact]"List data types.
dtype
string serialization
list[bt.CellType]"list[cat[bionty.CellType]]"list[float]"list[float]"Union data types.
Unions are currently only supported for static registries.
dtype
string serialization
[bt.Tissue.ontology_id, bt.CellType.ontology_id]"cat[bionty.Tissue.ontology_id|bionty.CellType.ontology_id]"Attributes¶
- property dtype_as_object: type | SQLRecord | DeferredAttribute | None¶
The
dtypeas an object.Example
For simple dtypes, returns the built-in Python type:
feature_float = ln.Feature(name="measurement", dtype=float).save() assert feature_float.dtype_as_object is float
For features with with
RecordorULabeltypes, returns theRecordorULabelobject:sample_type = bt.Record(name="Sample", is_type=True).save() feature_sample = ln.Feature(name="sample", dtype=sample_type).save() assert feature_sample.dtype_as_object == sample_type
For features with
Registrytypes, returns theRegistryobject or a field (DeferredAttribute) object:feature_cell_type = ln.Feature(name="cell_type_name", dtype=bt.CellType).save() assert feature_cell_type.dtype_as_object == bt.CellType feature_ontology_id = ln.Feature(name="ontology_id", dtype=bt.CellType.ontology_id).save() assert feature_ontology_id.dtype_as_object == bt.CellType.ontology_id
- property dtype_as_str: Literal['num', 'int', 'float', 'str', 'bool', 'datetime', 'datetime64[ns, UTC]', 'date', 'dict', 'path', 'url', 'object'] | str | None¶
The
dtypeas a string.You can query by this property as if it was a string field. The query is delegated to the private
_dtype_strfield.Is
NoneifFeatureifis_type=True, otherwise a string.Examples
Query by
dtype_as_str:ln.Feature.filter(dtype_as_str="float").to_dataframe()
Examples for
dtype_as_str:feature_float = ln.Feature(name="measurement", dtype=float).save() assert feature_float.dtype_as_str == "float" sample_type = bt.Record(name="Sample", is_type=True).save() feature_sample = ln.Feature(name="sample", dtype=sample_type).save() assert feature_sample.dtype_as_str == "cat[Record[12345678abcdeFGHI]] # uid of type record feature_list_float = ln.Feature(name="numbers", dtype=list[float]).save() assert feature_list_float.dtype_as_str == "list[float]" feature_ulabel = ln.Feature(name="sample", dtype=ln.ULabel).save() assert feature_ulabel.dtype_as_str == "cat[ULabel]" feature_record = ln.Feature(name="sample", dtype=bt.CellLine).save() assert feature_record.dtype_as_str == "cat[bionty.CellLine]" feature_list_record = ln.Feature(name="cell_types", dtype=list[bt.CellLine]).save() assert feature_list_record.dtype_as_str == "list[cat[bionty.CellLine]]"
- property notes: str | None¶
Notes.
Returns the latest content of an attached block of kind
readme.You can populate it via the UI or via
lamin annotate ... --readme README.md.
- property settings: SQLRecordSettings¶
Settings.
Simple fields¶
- uid: str¶
Universal id, valid across DB instances.
- name: str¶
Name of feature.
- unit: str | None¶
Unit of measure, ideally SI (
m,s,kg, etc.) or ‘normalized’ etc. (optional).
- description: str | None¶
A description.
- array_rank: int¶
Rank of feature.
Number of indices of the array: 0 for scalar, 1 for vector, 2 for matrix.
Is called
.ndiminnumpyandpytorchbut shouldn’t be confused with the dimension of the feature space.
- array_size: int¶
Number of elements of the feature.
Total number of elements (product of shape components) of the array.
A number or string (a scalar): 1 or
NoneA 50-dimensional embedding: 50
A 25 x 25 image: 625
- array_shape: list[int] | None¶
Shape of the feature.
A number or string (a scalar): [1] or
NoneA 50-dimensional embedding: [50]
A 25 x 25 image: [25, 25]
Is stored as a list rather than a tuple because it’s serialized as JSON.
- synonyms: str | None¶
Bar-separated (|) synonyms (optional).
- default_value: Any | None¶
A default value that overwrites missing values during standardization.
- nullable: bool | None¶
Whether the feature can have nullable values. None for type-like features.
- coerce: bool | None¶
Whether dtypes should be coerced during validation. None for type-like features.
- is_type: bool¶
Indicates if record is a
type.For example, if a record “Compound” is a
type, the actual compounds “darerinib”, “tramerinib”, would be instances of thattype.
- is_locked: bool¶
Whether the object is locked for edits.
- created_at: datetime¶
Time of creation of record.
- updated_at: datetime¶
Time of last update to record.
Relational fields¶
- type: Feature | None¶
Type of feature (e.g., ‘Readout’, ‘Metric’, ‘Metadata’, ‘ExpertAnnotation’, ‘ModelPrediction’).
Allows to group features by type, e.g., all read outs, all metrics, etc.
- schemas: RelatedManager[Schema]¶
Schemas linked to this feature.
- features: RelatedManager[Feature]¶
Features of this type (can only be non-empty if
is_typeisTrue).
- values: RelatedManager[JsonValue]¶
Values for this feature.
- projects: RelatedManager[Project]¶
Annotating projects.
- ablocks: RelatedManager[FeatureBlock]¶
Attached blocks ←
feature.
Class methods¶
- classmethod from_dataframe(df, field=None, *, mute=False)¶
Create Feature records for dataframe columns.
- Parameters:
df (
DataFrame) – Source DataFrame to extract column information fromfield (
DeferredAttribute|None, default:None) – FieldAttr for Feature model validation, defaults to Feature.namemute (
bool, default:False) – Whether to mute Feature creation similar names found warnings
- Return type:
SQLRecordList
- classmethod from_dict(dictionary, field=None, *, type=None, mute=False)¶
Create Feature records for dictionary keys.
- Parameters:
dictionary (
dict[str,Any]) – Source dictionary to extract key information fromfield (
DeferredAttribute|None, default:None) – FieldAttr for Feature model validation, defaults toFeature.nametype (
Feature|None, default:None) – Feature type of all created featuresmute (
bool, default:False) – Whether to mute dtype inference and feature creation warnings
- Return type:
SQLRecordList
- classmethod filter(*queries, **expressions)¶
Query records.
- Parameters:
queries – One or multiple
Qobjects.expressions – Fields and values passed as Django query expressions.
- Return type:
See also
Guide: Query & search
Django documentation: Queries
Examples
>>> ln.Project(name="my label").save() >>> ln.Project.filter(name__startswith="my").to_dataframe()
- classmethod get(idlike=None, **expressions)¶
Get a single record.
- Parameters:
idlike (
int|str|None, default:None) – Either a uid stub, uid or an integer id.expressions – Fields and values passed as Django query expressions.
- Raises:
lamindb.errors.ObjectDoesNotExist – In case no matching record is found.
- Return type:
See also
Guide: Query & search
Django documentation: Queries
Examples
record = ln.Record.get("FvtpPJLJ") record = ln.Record.get(name="my-label")
- classmethod to_dataframe(include=None, features=False, limit=100)¶
Evaluate and convert to
pd.DataFrame.By default, this returns up to 20 rows for a fast overview. Pass
limit=Noneto fetch all matching records.By default, maps simple fields and foreign keys onto
DataFramecolumns.Guide: Query & search
- Parameters:
include (
str|list[str] |None, default:None) – Related data to include as columns. Takes strings of form"records__name","cell_types__name", etc. or a list of such strings. ForArtifact,Record, andRun, can also pass"features"to include features measured in the current queryset. If"privates", includes private fields (fields starting with_).features (
bool|list[str], default:False) – Configure the features to include. Can be a feature name or a list of such names. Only available forArtifact,Record, andRun.limit (
int, default:100) – Maximum number of rows to display. Defaults to 20. IfNone, includes all results.order_by – Field name to order the records by. Prefix with ‘-’ for descending order. Defaults to ‘-id’ to get the most recent records. This argument is ignored if the queryset is already ordered or if the specified field does not exist.
- Return type:
DataFrame
Examples
Include the name of the creator:
ln.Record.to_dataframe(include="created_by__name"])
Include features:
ln.Artifact.to_dataframe(include="features")
Include selected features:
ln.Artifact.to_dataframe(features=["cell_type_by_expert", "cell_type_by_model"])
- classmethod search(string, *, field=None, limit=20, case_sensitive=False)¶
Search.
- Parameters:
string (
str) – The input string to match against the field ontology values.field (
str|DeferredAttribute|None, default:None) – The field or fields to search. Search all string fields by default.limit (
int|None, default:20) – Maximum amount of top results to return.case_sensitive (
bool, default:False) – Whether the match is case sensitive.
- Return type:
- Returns:
A sorted
DataFrameof search results with a score in columnscore. Ifreturn_querysetisTrue.QuerySet.
See also
filter()lookup()Examples
records = ln.ULabel.from_values(["Label1", "Label2", "Label3"]).save() ln.ULabel.search("Label2")
- classmethod lookup(field=None, return_field=None)¶
Return an auto-complete object for a field.
- Parameters:
field (
str|DeferredAttribute|None, default:None) – The field to look up the values for. Defaults to first string field.return_field (
str|DeferredAttribute|None, default:None) – The field to return. IfNone, returns the whole record.keep – When multiple records are found for a lookup, how to return the records. -
"first": return the first record. -"last": return the last record. -False: return all records.
- Return type:
NamedTuple- Returns:
A
NamedTupleof lookup information of the field values with a dictionary converter.
See also
search()Examples
Lookup via auto-complete on
.:import bionty as bt bt.Gene.from_source(symbol="ADGB-DT").save() lookup = bt.Gene.lookup() lookup.adgb_dt
Look up via auto-complete in dictionary:
lookup_dict = lookup.dict() lookup_dict['ADGB-DT']
Look up via a specific field:
lookup_by_ensembl_id = bt.Gene.lookup(field="ensembl_gene_id") genes.ensg00000002745
Return a specific field value instead of the full record:
lookup_return_symbols = bt.Gene.lookup(field="ensembl_gene_id", return_field="symbol")
- classmethod connect(instance)¶
Query a non-default LaminDB instance.
- Parameters:
instance (
str|None) – An instance identifier of form “account_handle/instance_name”.- Return type:
Examples
ln.Record.connect("account_handle/instance_name").search("label7", field="name")
- classmethod inspect(field=None, *, mute=False, organism=None, source=None, from_source=True, strict_source=False)¶
Inspect if values are mappable to a field.
Being mappable means that an exact match exists.
- Parameters:
values (
Sequence[str]) – Values that will be checked against the field.field (
str|DeferredAttribute|None, default:None) – The field of values. Examples are'ontology_id'to map against the source ID or'name'to map against the ontologies field names.mute (
bool, default:False) – Whether to mute logging.organism (
str|SQLRecord|None, default:None) – An Organism name or record.source (
SQLRecord|None, default:None) – Abionty.Sourcerecord that specifies the version to inspect against.strict_source (
bool, default:False) – Determines the validation behavior against records in the registry. - IfFalse, validation will include all records in the registry, ignoring the specified source. - IfTrue, validation will only include records in the registry that are linked to the specified source. Note: this parameter won’t affect validation against public sources.
- Return type:
bionty.base.dev.InspectResult
See also
Example
Inspect gene symbols:
import bionty as bt # populate the gene registry bt.Gene.from_values(["A1CF", "A1BG", "BRCA2"], field="symbol", organism="human").save() # inspect gene symbols symbols = ["A1CF", "A1BG", "FANCD1", "FANCD20"] result = bt.Gene.inspect(symbols, field=bt.Gene.symbol, organism="human") assert result.validated == ["A1CF", "A1BG"] assert result.non_validated == ["FANCD1", "FANCD20"]
- classmethod validate(field=None, *, mute=False, organism=None, source=None, strict_source=False)¶
Validate values against existing values of a string field.
Note this is strict_source validation, only asserts exact matches.
- Parameters:
values (
Sequence[str]) – Values that will be validated against the field.field (
str|DeferredAttribute|None, default:None) – The field of values. Examples are'ontology_id'to map against the source ID or'name'to map against the ontologies field names.mute (
bool, default:False) – Whether to mute logging.organism (
str|SQLRecord|None, default:None) – An Organism name or record.source (
SQLRecord|None, default:None) – Abionty.Sourcerecord that specifies the version to validate against.strict_source (
bool, default:False) – Determines the validation behavior against records in the registry. - IfFalse, validation will include all records in the registry, ignoring the specified source. - IfTrue, validation will only include records in the registry that are linked to the specified source. Note: this parameter won’t affect validation against public sources.
- Return type:
ndarray- Returns:
A vector of booleans indicating if an element is validated.
See also
Example
Validate gene symbols:
import bionty as bt # populate the gene registry bt.Gene.from_values(["A1CF", "A1BG", "BRCA2"], field="symbol", organism="human").save() # validate gene symbols symbols = ["A1CF", "A1BG", "FANCD1", "FANCD20"] bt.Gene.validate(symbols, field=bt.Gene.symbol, organism="human") #> array([ True, True, False, False])
- classmethod from_values(field=None, create=False, organism=None, source=None, mute=False)¶
Bulk create validated records by parsing values for an identifier such as a name or an id).
- Parameters:
values (
Sequence[str]) – A list of values for an identifier, e.g.["name1", "name2"].field (
str|DeferredAttribute|None, default:None) – ASQLRecordfield to look up, e.g.,bt.CellMarker.name.create (
bool, default:False) – Whether to create records if they don’t exist.organism (
SQLRecord|str|None, default:None) – Abionty.Organismname or record.source (
SQLRecord|None, default:None) – Abionty.Sourcerecord to validate against to create records for.mute (
bool, default:False) – Whether to mute logging.
- Return type:
SQLRecordList- Returns:
A list of validated records. For bionty registries. Also returns knowledge-coupled records.
Notes
For more info, see tutorial: Manage biological ontologies.
Example
Bulk create labels:
# from invalid values logs warnings & returns an empty list ulabels = ln.ULabel.from_values(["benchmark", "prediction", "test"]) assert len(ulabels) == 0 # from valid values or via `create=True` returns label objects ulabels = ln.ULabel.from_values(["benchmark", "prediction", "test"], create=True).save() assert len(ulabels) == 3 # bulk create cell type labels from a public ontology import bionty as bt bt.CellType.from_values(["T cell", "B cell"]).save()
- classmethod standardize(field=None, *, return_field=None, return_mapper=False, case_sensitive=False, mute=False, from_source=True, keep='first', synonyms_field='synonyms', organism=None, source=None, strict_source=False)¶
Maps input synonyms to standardized names.
- Parameters:
values (
Sequence[str]) – Identifiers that will be standardized.field (
str|DeferredAttribute|None, default:None) – The field representing the standardized names.return_field (
str|DeferredAttribute|None, default:None) – The field to return. Defaults to field.return_mapper (
bool, default:False) – IfTrue, returns{input_value: standardized_name}.case_sensitive (
bool, default:False) – Whether the mapping is case sensitive.mute (
bool, default:False) – Whether to mute logging.from_source (
bool, default:True) – Whether to standardize from public source. Defaults toTruefor BioRecord registries.keep (
Literal['first','last',False], default:'first') –When a synonym maps to multiple names, determines which duplicates to mark as
pd.DataFrame.duplicated: -"first": returns the first mapped standardized name -"last": returns the last mapped standardized name -False: returns all mapped standardized name.When
keepisFalse, the returned list of standardized names will contain nested lists in case of duplicates.When a field is converted into return_field, keep marks which matches to keep when multiple return_field values map to the same field value.
synonyms_field (
str, default:'synonyms') – A field containing the concatenated synonyms.organism (
str|SQLRecord|None, default:None) – An Organism name or record.source (
SQLRecord|None, default:None) – Abionty.Sourcerecord that specifies the version to validate against.strict_source (
bool, default:False) – Determines the validation behavior against records in the registry. - IfFalse, validation will include all records in the registry, ignoring the specified source. - IfTrue, validation will only include records in the registry that are linked to the specified source. Note: this parameter won’t affect validation against public sources.
- Return type:
list[str] |dict[str,str]- Returns:
If
return_mapperisFalse– a list of standardized names. Otherwise, a dictionary of mapped values with mappable synonyms as keys and standardized names as values.
See also
add_synonym()Add synonyms.
remove_synonym()Remove synonyms.
Example
Standardize gene identifiers:
import bionty as bt # save some gene objects bt.Gene.from_values(["A1CF", "A1BG", "BRCA2"], field="symbol", organism="human").save() # standardize gene synonyms gene_synonyms = ["A1CF", "A1BG", "FANCD1", "FANCD20"] bt.Gene.standardize(gene_synonyms) #> ['A1CF', 'A1BG', 'BRCA2', 'FANCD20']
Methods¶
- is_null(value=True)¶
Build a predicate for null-style feature presence checks.
Example:
perturbation = ln.Feature.get(name="perturbation") ln.Artifact.filter(perturbation.is_null(False)).to_dataframe( include="features" )
- Return type:
FeaturePredicate
- query_features()¶
Query features of sub types.
While
.featuresretrieves the features with the current type, this method also retrieves sub types and the features with sub types of the current type.- Return type:
- with_config(optional=None)¶
Pass addtional configurations to the schema.
- Return type:
tuple[Feature,dict]
- restore()¶
Restore from trash onto the main branch.
Does not restore descendant objects if the object is
HasTypewithis_type = True.- Return type:
None
- delete(permanent=None, **kwargs)¶
Delete object.
If object is
HasTypewithis_type = True, deletes all descendant objects, too.- Parameters:
permanent (
bool|None, default:None) – Whether to permanently delete the object (skips trash). IfNone, performs soft delete if the object is not already in the trash.- Returns:
When
permanent=True, returns Django’s delete return value – a tuple of (deleted_count, {registry_name: count}). Otherwise returns None.
Examples
For any
SQLRecordobjectsqlrecord, call:sqlrecord.delete()
- classmethod describe(include=None)¶
Describe record including relations.
- Parameters:
return_str (
bool, default:False) – Return a string instead of printing.include (
None|Literal['comments'], default:None) – Include additional content. Use"comments"to display readme and comment blocks.
- Return type:
None|str
- query_types()¶
Query types of a record recursively.
While
.typeretrieves thetype, this method retrieves all super types of thattype:# Create type hierarchy type1 = model_class(name="Type1", is_type=True).save() type2 = model_class(name="Type2", is_type=True, type=type1).save() type3 = model_class(name="Type3", is_type=True, type=type2).save() # Create a record with type3 record = model_class(name=f"{model_name}3", type=type3).save() # Query super types super_types = record.query_types() assert super_types[0] == type3 assert super_types[1] == type2 assert super_types[2] == type1
- Return type:
SQLRecordList
- add_synonym(synonym, force=False, save=None)¶
Add synonyms to a record.
- Parameters:
synonym (
str|Sequence[str]) – The synonyms to add to the record.force (
bool, default:False) – Whether to add synonyms even if they are already synonyms of other records.save (
bool|None, default:None) – Whether to save the record to the database.
See also
remove_synonym()Remove synonyms.
Example
Add a synonym for a cell type:
import bionty as bt # create a "T cell" object t_cell = bt.CellType.from_source(name="T cell").save() t_cell.synonyms #> "T-cell|T lymphocyte|T-lymphocyte" # add a synonym t_cell.add_synonym("T cells") t_cell.synonyms #> "T cells|T-cell|T-lymphocyte|T lymphocyte"
- remove_synonym(synonym)¶
Remove synonyms from a record.
- Parameters:
synonym (
str|Sequence[str]) – The synonym values to remove.
See also
add_synonym()Add synonyms
Example
Remove a synonym for a cell type:
import bionty as bt # save "T cell" record record = bt.CellType.from_source(name="T cell").save() record.synonyms #> "T-cell|T lymphocyte|T-lymphocyte" # remove a synonym record.remove_synonym("T-cell") record.synonyms #> "T lymphocyte|T-lymphocyte"
- refresh_from_db(using=None, fields=None, from_queryset=None)¶
Reload field values from the database.
By default, the reloading happens from the database this instance was loaded from, or by the read router if this instance wasn’t loaded from any database. The using parameter will override the default.
Fields can be used to specify which fields to reload. The fields should be an iterable of field attnames. If fields is None, then all non-deferred fields are reloaded.
When accessing deferred fields of an instance, the deferred loading of the field will call this method.